
非贵金属三元复合Ni(PO3)2-Ni2P/CdS NPs异质结的构建及可见光高效催化产氢性能
2022-07-18 02:41:20王广琦毕艺洋王嘉博石洪飞刘群张钰
高等学校化学学报 2022年6期
王广琦, 毕艺洋, 王嘉博, 石洪飞, 刘群, 张钰
非贵金属三元复合Ni(PO3)2-Ni2P/CdS NPs异质结的构建及可见光高效催化产氢性能
王广琦1, 毕艺洋2, 王嘉博2, 石洪飞2, 刘群2, 张钰2
(1. 吉林化工学院材料科学与工程学院, 2. 石油化工学院, 吉林 132022)
结合异质结构建与共催化剂改性, 以花球状Ni(OH)2为前驱体, 经热磷酸化后得到Ni(PO3)2-Ni2P二元助催化剂, 借助超声化学合成法, 与CdS NPs复合, 形成非贵金属CdS基三元光催化材料Ni(PO3)2-Ni2P/CdS NPs. 以Na2S-Na2SO3为牺牲剂, 在可见光(>420 nm)照射下, 在不借助任何贵金属的情况下, 负载量为8%(质量分数)的Ni(PO3)2-Ni2P/CdS NPs复合材料的光催化产氢速率达到4237 μmol·g‒1·h‒1, 为CdS NPs(217 μmol·g‒1·h‒1)的19倍. 在产氢循环实验中, 反应进行到第6次循环(18 h)后, 复合材料的产氢速率约为初始的89%, 具有较好的稳定性. 与CdS NPs相比, Ni(PO3)2-Ni2P/CdS NPs的吸收边明显红移, 禁带宽度降至1.86 eV, 并降低了H+还原的过电位, 显示出增强的光吸收性能和适宜的带隙结构. 通过Ni(PO3)2-Ni2P与CdS NPs之间的协同效应, 有效促进了光生载流子的分离, 提高了产氢活性和稳定性.
硫化镉纳米粒子; 偏磷酸镍; 磷化镍; 光催化; 产氢
新能源开发是21世纪亟待实现的首要目标[1]. 氢能作为一种高效、 清洁及可持续的 “无碳” 能源, 被公认为是最理想的化石能源替代者[2,3]. 同时, 太阳能作为一种天然存在的新能源, 已被认为是最有前景的清洁能源之一[4]. 低密度的太阳能通过光催化技术可以转化为高密度的化学能, 如光解水产氢[5]. 通过太阳光驱动半导体材料催化光解水产氢已引起广泛关注[6]. 涉及多学科方法的光催化分解水产氢催化剂已被报道, 如非金属化合物(g-C3N4[7~9]、 石墨烯[10,11])、 金属硫化物(CdS[12~14], MoS2[15,16], ZnS[17,18])、 金属氧化物(TiO2[19,20], ZnO[21])和金属氮氧化物(TaON[22])等. 对于常用的宽禁带半导体而言, 金属硫化物半导体通常拥有较窄的带隙, 可以在更大程度上有效利用太阳能[23]. 在金属硫化物半导体中, CdS半导体拥有较负的导带位置和较窄的带隙(2.4 eV), 相应的本征光吸收带边为517 nm, 是一种可见光响应型半导体, 在太阳能转化领域受到了广泛关注[24]. 然而, CdS在溶液中经过长时间光照射后, 光腐蚀现象严重, 并表现出样品不稳定的趋势, 而且自身载流子极容易复合, 限制了其在光催化方面的应用[25]. 因此, 选择适宜的改性方法, 增强CdS光吸收性能、 拓宽光吸收范围、 抑制光生载流子的复合, 从而提高光催化活性及稳定性, 对提升CdS光催化性能具有重大意义[26].
增强CdS半导体光催化性能的策略主要有元素掺杂[27]、 形貌调控[28]、 构建异质结和助剂负载[29]等.贵金属或非贵金属还原助催化剂的引入, 是改良半导体材料光解水产氢的有效方式, 在提升材料吸光能力、 加速电子迁移和抑制载流子复合等方面均有显著作用[30]. 常用的还原助催化剂有Pt, Au, Ru和Rh等贵金属, 以及部分硫化物(如MoS2)和磷化物(如NiP)等[31,32]. 然而, 贵金属的高成本和低丰度大大限制了它们的广泛应用[33]. 在非贵金属还原助催化剂中, 镍基助催化剂因成本低、 活性高和稳定性好而备受关注[34]. 已有大量研究表明, 磷化镍与半导体材料复合, 作为助催化剂可以有效提高半导体的产氢效率[35]. Wu等[36]研究发现, 硫化镉纳米棒上的g-C3N4可以有效促进电子-空穴对的空间分离, 磷化镍(Ni2P)可以降低H+还原的过电位. 此外, Ni2P 助催化剂的表面负载能够进一步从涂有碳层(CdS@C) 异质结的硫化镉中吸引光生电子, 并为还原产氢提供活性位点[37]. 然而, 由于Ni2P的催化位点密度不足, 催化活性仍受限制[38].
近年来, 镍离子和磷酸根也逐渐进入了光催化研究领域. 金属磷酸盐体系倾向于模仿天然酶催化过程的基本原理, 其中官能团之间的静电相互作用和氢键等弱相互作用对电荷的分离起到协同作用[39]. 相比于磷化镍, 各类磷酸镍具有更低的带隙, 可增大光谱吸收范围, 具有提高光催化效率的潜力[40]. Samal等[41]基于磷酸镍良好的耐热性、 耐光性和强吸收性, 首次制备了还原氧化石墨烯/磷酸镍(RGO-NiPO)复合材料, 并用于高效光催化产氢. Jiang等[42]采用水热法和煅烧法, 制备了Ni2P/ Ni(PO3)2/g-C3N4三元三维异质结光催化剂, 通过负载非贵金属磷化物和偏磷酸盐混合物Ni2P/Ni(PO3)2, 可以提高g-C3N4光催化剂的光催化性能. Gurbani等[43]采用原位太阳能辅助一锅法, 制备了氧化石墨 烯@磷酸镍(GO∶NPO)纳米复合材料, 光生载流子通过界面实现空间分离, 光致电子从 NiPO4转移到GO片, 从而抑制了电荷空穴复合. 目前, 磷酸镍作为光催化产氢助催化剂的研究尚处于起步阶段, 深入研究其助催化性能及机理, 为光催化产氢领域开发更高效的非贵金属助催化剂具有重要意义.
基于此, 本文将结晶Ni(PO3)2薄层和Ni2P纳米粒子作为共催化剂, 与硫化镉纳米粒子(CdS NPs) 紧密接触形成异质结构, 合成了以Ni(PO3)2-Ni2P二元材料为非贵金属助催化剂的CdS基三元复合光催化剂Ni(PO3)2-Ni2P/CdS NPs, 通过荧光光谱、 电化学阻抗谱和瞬态光电流响应谱等方法, 研究了 Ni(PO3)2-Ni2P和CdS NPs之间光生载流子的有效转移和分离, 并研究了Ni(PO3)2-Ni2P/CdS NPs的可见光催化产氢性能.
1 实验部分
1.1 试剂与仪器
二水合乙酸镉 [Cd(OAC)2·2H2O]、 九水合硫化钠(Na2S·9H2O)、 聚乙烯吡咯烷酮(PVP)、 六水合硝酸镍[Ni(NO3)2·6H2O]、 次亚磷酸钠(NaH2PO2)、 乌洛托品(C6H12N4)和亚硫酸钠(Na2SO3)均为分析纯, 购自上海阿拉丁试剂公司; 无水乙醇(C2H6O, 分析纯)购自天津化学试剂厂.
D8-ADVANCE型X射线衍射仪(XRD, 德国布鲁克公司); JSM-7610FPlus型扫描电子显微镜(SEM, 日本电子株式会社); Sigma 500型场发射扫描电子显微镜(FESEM, 德国卡尔蔡司光学有限公司); Nicolet is10型傅里叶变换红外光谱仪(FTIR)和Escalab 250Xi型X射线光电子能谱(XPS)(美国赛默飞世尔科技公司); Lambda750型紫外-可见吸收光谱仪(UV-Vis)和ls55型荧光分光光度计(PL)(美国珀金埃尔默仪器公司); CHI-760E型电化学工作站(上海辰华公司); GC-2014型气相色谱仪(GC, 日本岛津公司).
1.2 光催化剂的制备
1.2.1CdS NPs的制备将0.16 g PVP加入200 mL 0.04 mol/L Cd(OAC)2·2H2O水溶液中, 并在剧烈搅拌下滴加200 mL 0.04 mol/L Na2S水溶液. 使用微孔滤膜过滤所得橙黄色悬浊液, 分别用无水乙醇和去离子水依次洗涤各3次, 在真空烘箱中于60 ℃干燥8 h, 得到 CdS 纳米粒子, 记为CdS NPs.
1.2.2Ni(PO3)2-Ni2P的制备将2 g Ni(NO3)2·6H2O和2 g C6H12N4加入10 mL去离子水中并搅拌2 h[44]. 然后, 将溶液转移到20 mL内衬为聚四氟乙烯的反应釜中, 在120 ℃下水热反应12 h. 所得产物经离心分离后, 在真空烘箱中于60 ℃干燥8 h得到Ni(OH)2. 将0.2 g Ni(OH)2和2 g NaH2PO2分别置于瓷舟中, 并在25 ℃下顺次置于内径为50 mm的石英管式炉中, 且装有NaH2PO2的瓷舟靠近进气端. 在20 mL/min氩气下, 以5 ℃/min的升温速率加热到350 ℃并恒温磷化, 即可得到黑色固体粉末Ni(PO3)2-Ni2P; 通过控制恒温时间, 调节Ni(PO3)2与Ni2P的比例, 热磷酸化2 h的样品标记为Ni(PO3)2-Ni2P-1, 热磷酸化3 h的样品标记为Ni(PO3)2-Ni2P-2.
1.2.3Ni(PO3)2-Ni2P/CdS NPs的制备将0.017 g Ni(PO3)2-Ni2P-1分散在30 mL乙醇中, 并超声处理 0.5 h. 然后, 加入0.2 g CdS NPs继续超声20 min. 将所得悬浮液旋转蒸发后, 置于真空干燥箱中于60 ℃下干燥8 h, 得到 Ni(PO3)2-Ni2P-1质量分数为8% 的Ni(PO3)2-Ni2P-1/CdS NPs; 按相同方法制备了8% 的Ni(PO3)2-Ni2P-2/CdS NPs, 以用于Ni(PO3)2与Ni2P不同比例的催化效果考察. 此外, 为了研究 Ni(PO3)2-Ni2P与CdS NPs的适宜比例, 分别制备了Ni(PO3)2-Ni2P-1质量分数分别为1%, 3%, 5%和10%的复合催化剂. 催化剂制备过程如Scheme 1所示.

Scheme 1Schematic illustration for the preparation of Ni(PO3)2⁃Ni2P(A) and Ni(PO3)2⁃Ni2P/CdS NPs(B)
1.3 光催化活性测试
将50 mg光催化剂分散于装有50 mL 0.35 mol/L Na2S和0.25 mol/L Na2SO3混合溶液的顶照式Pyrex石英光催化反应器中, 采用低温循环水机保持光催化活性测试温度为10 ℃. 氙灯光源(>420 nm, 300 W)与光催化反应器的距离调整保持在3 cm, 在磁力搅拌下进行液固相光催化反应.每间隔1 h用气相色谱仪和热导检测器(TCD)进行定量分析.
2 结果与讨论
2.1 光催化材料的表征


Fig.1 XRD patterns(A) and FTIR spectra(B) of Ni(OH)2(a), Ni(PO3)2⁃Ni2P⁃1(b) and Ni(PO3)2⁃Ni2P⁃2(c)
图1(B)示出了-Ni(OH)2和Ni(PO3)2-Ni2P的红外光谱图.-Ni(OH)2, Ni(PO3)2-Ni2P-1和Ni(PO3)2-Ni2P-2均在3400 cm‒1处出现很宽的羟基振动吸收峰, 其中羟基可能来自于残留的水, 或者与镍或磷结合的羟基, 或者羟基之间产生的氢键的吸收峰.-Ni(OH)2在1650和1463 cm‒1附近的峰代表水分子的弯曲振动, 而在654.7 和490.3 cm‒1附近的峰分别与Ni—O伸缩振动和面内Ni—O—H弯曲振动有关[47]. 另外,-Ni(OH)2在1382 cm‒1处的强峰说明硝酸盐阴离子的存在, 1011和1248 cm‒1处的特征峰归属于C—N对称和反对称伸缩振动, 821 cm‒1处特征峰归属于C—N弯曲振动, 说明乌洛托品存在残留. Ni(PO3)2-Ni2P-1和Ni(PO3)2-Ni2P-2在765.7, 1044.8, 1236.6和1549.7 cm‒1处的吸收峰可分别 归属于s(POP),as(POP),s(PO2)和as(PO2)的伸缩振动, 在526.0 和2350.0 cm‒1处的吸收峰归属于(Ni—O)伸缩振动[48]. 以上分析表明,-Ni(OH)2已经转化为Ni(PO3)2-Ni2P, Ni(PO3)2-Ni2P-1和 Ni(PO3)2-Ni2P-2中均存在Ni(PO3)2和Ni2P的组分.
采用SEM分析了样品的表观形貌[图2(A)~(F)]. 图2(A)和(B)分别为Ni(OH)2及局部放大的SEM照片. 可见, Ni(OH)2是由纳米薄层交叉团聚而成的花球状形貌, 纳米层片分散良好, 厚度均匀(约10 nm), 花球直径范围为3~5 µm. 将Ni(OH)2热磷酸化2 h后, 得到的Ni(PO3)2-Ni2P-1呈现层状晶片堆积形貌[图2(C)和(D)], 层片厚度约200 nm, 在层片表面及层间可见100 nm左右的Ni2P晶粒随机分布. Ni(OH)2热磷酸化3 h后, 得到的Ni(PO3)2-Ni2P-2晶片层坍塌严重, 更倾向于粉化的状态, 可能是Ni2P增多的原因所导致[图2(E)和(F)]. 表明短时间热磷化主要生成Ni(PO3)2, 长时间磷化会逐步脱氧生成Ni2P. 此外, 分别进行了Ni(PO3)2-Ni2P-1和Ni(PO3)2-Ni2P-2的X射线能谱(EDX)元素分析[图S1、 表S1和表S2, 见本文支持信息], 经计算, Ni(PO3)2-Ni2P-1中Ni(PO3)2和Ni2P的质量比是1.36∶1, 以 Ni(PO3)2为主; Ni(PO3)2-Ni2P-2中Ni(PO3)2和Ni2P的质量比是0.59∶1, Ni2P含量大幅提高; 与XRD, FTIR和SEM的表征结果一致. 短时间热磷酸化主要得到Ni(PO3)2, 长时间热磷酸化后Ni2P含量增加.

Fig.2 SEM images of Ni(OH)2(A, B), Ni(PO3)2⁃Ni2P⁃1(C, D) and Ni(PO3)2⁃Ni2P⁃2(E ,F)

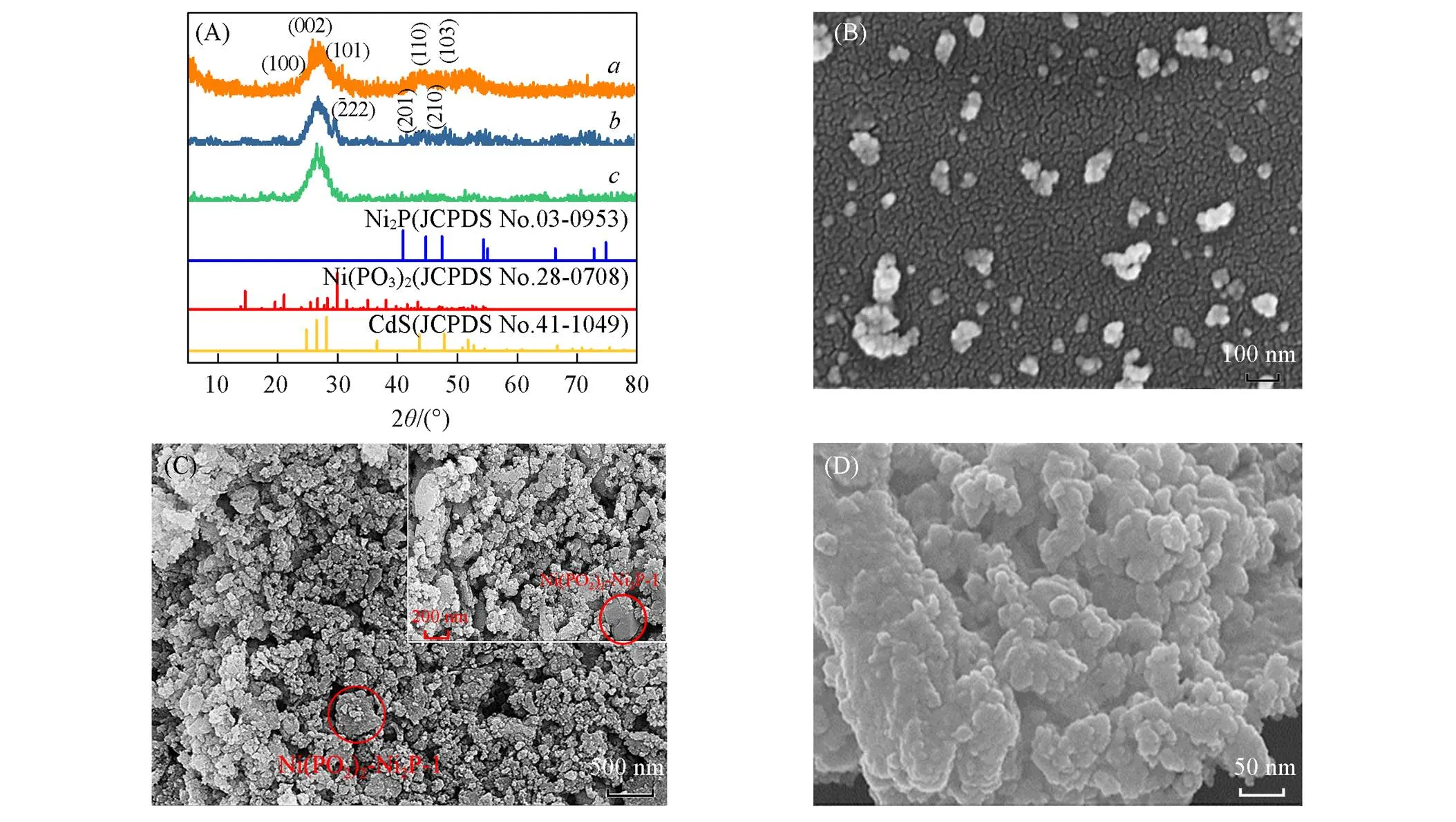
Fig.3 XRD patterns of CdS NPs(a), 8%Ni(PO3)2⁃Ni2P⁃1/CdS NPs(b) and 8%Ni(PO3)2⁃Ni2P⁃2/CdS NPs(c)(A), SEM images of CdS NPs(B), 8%Ni(PO3)2⁃Ni2P⁃1/CdS NPs(C) and 8%Ni(PO3)2⁃Ni2P⁃2/CdS NPs(D)
采用SEM分析了复合催化剂样品的表观形貌[图3(B)~(D)]. 由图3(B)可见, 通过PVP辅助常温沉淀制备的CdS为纳米颗粒(10~30 nm)形貌, 且分散良好. 8%Ni(PO3)2-Ni2P-1/CdS NPs的SEM照片表明, 超声处理后CdS NPs均匀地分散于Ni(PO3)2-Ni2P-1层片上[图3(C)]. 催化剂的形貌是影响光催化活性的因素之一, 纳米薄层结构具有表面积大、 径向距离短和电荷分离效率快等优点, 这可能有助于提高其光催化析氢的活性. 图3(D)展示了8%Ni(PO3)2-Ni2P-2/CdS NPs的形貌, 从图2(E)和(F)可以看出, Ni(PO3)2-Ni2P-2趋向于片层坍塌、 粉化, 复合过程中在超声空化的强作用下, Ni(PO3)2-Ni2P-2以纳米颗粒的状态与CdS NPs复合重整, 更有利于异质结的形成, 但是颗粒堆积包覆可能会影响CdS NPs的光吸收, 不如CdS NPs沉积在Ni(PO3)2-Ni2P-1片层上的光吸收性能好, 从而影响产氢性能. 另外, 采用EDX研究了8%Ni(PO3)2-Ni2P-1/CdS NPs的元素分布[图S2(A)~(E), 见本文支持信息] , 以及 8%Ni(PO3)2-Ni2P-1/CdS NPs和8%Ni(PO3)2-Ni2P-2/CdS NPs的元素组成[图S3(A)和(B), 见本文支持信息]. 复合材料中存在O, P, Ni, Cd和S元素, 各元素分布均匀, 以CdS NPs组分为主.
采用XPS测试了8%Ni(PO3)2-Ni2P-1/CdS NPs复合材料的化学组成和元素状态. 结果表明, 吸附的气体分子中存在Cd, S, Ni, P和O元素, 如图4(A)所示. Cd3dXPS谱在405.4和412.2 eV处有两个明显的峰[图4(B)], 分别对应于Cd3d5/2和Cd3d3/2, 表明Cd2+离子在剥离的CdS NPs中, 复合形成异质结后价态未发生变化[49]. 从图4(C)可以看到, 出现在161.5和162.7 eV处的两个峰分别归属于CdS NPs中的S2p3/2和S2p1/2[50]. 由图4(D)可见, 位于857.5和874.6 eV处的峰可以分别归属于Ni2+的Ni2p3/2和Ni2p1/2[51], 出现在863.6 eV处的峰指向Ni2p3/2卫星峰[52]. 由图4(E)可见, P2p的XPS光谱中出现在125.3 eV处的峰归属于Ni2P中的P2p3/2, 在133.3 eV处的峰可归属于P—O键, 对应于富磷条件下合成过程中催化剂表面的磷酸盐键合[53]. 由图4(F)可见, 位于533.1和531.1 eV处的峰对应于P—O—Ni和P—O—P键, 说明体系中存在Ni(PO3)2.
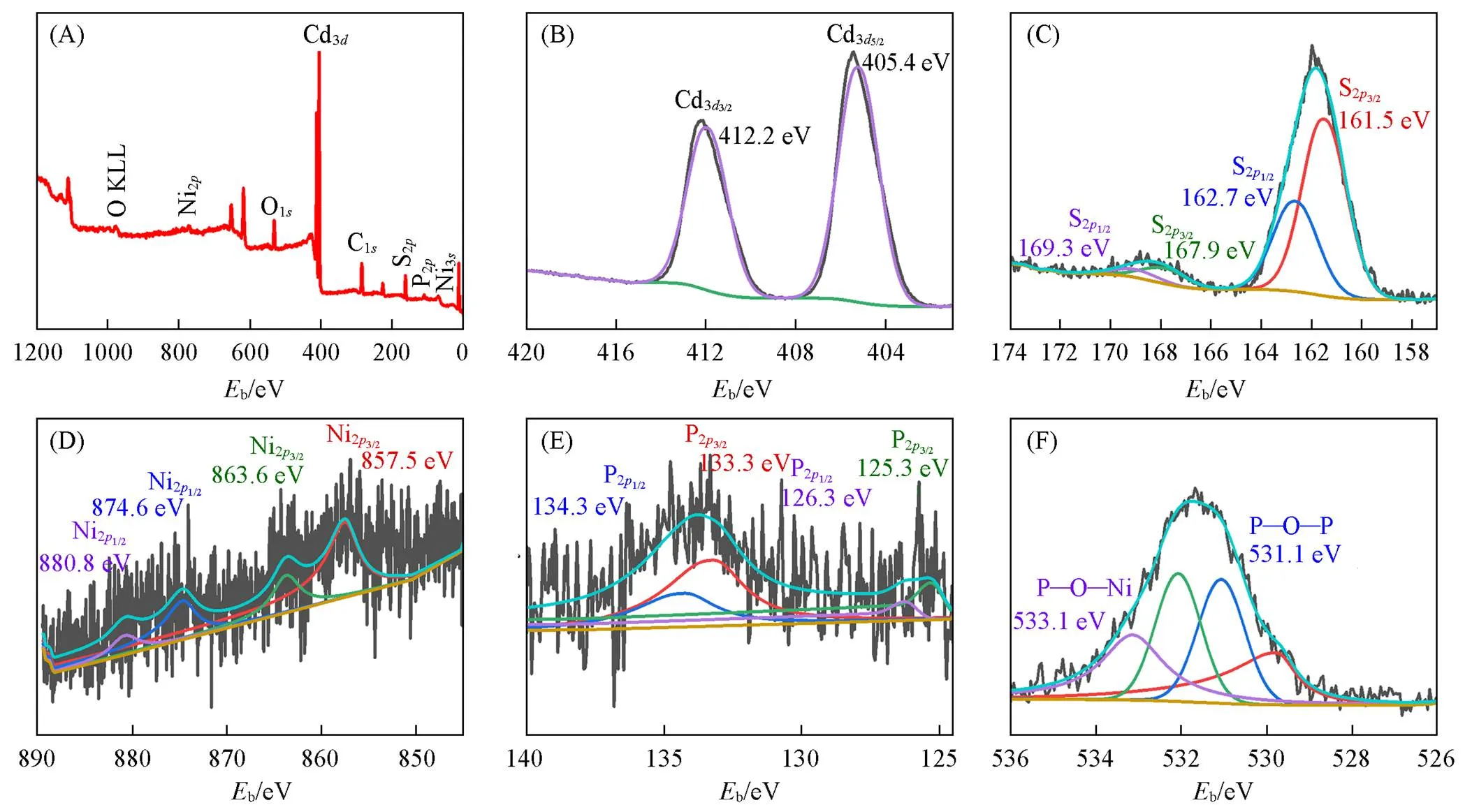
Fig.4 XPS spectra of 8%Ni(PO3)2⁃Ni2P⁃1/CdS NPs
(A) Full spectra; (B) Cd3d; (C) S2p; (D) Ni2p; (E) P2p; (F) O1s.
通常, 光催化活性主要依赖于催化材料对光的吸收能力和参与氧化还原反应离子的快速迁移以及电荷分离情况[54]. 图5(A)为CdS NPs, Ni(PO3)2-Ni2P-1, Ni(PO3)2-Ni2P-2, 8%Ni(PO3)2-Ni2P-1/CdS NPs和8%Ni(PO3)2-Ni2P-2/CdS NPs的紫外-可见漫反射光谱图. 可见, CdS NPs, Ni(PO3)2-Ni2P-1和 Ni(PO3)2-Ni2P-2的吸收带边分别为526, 621和573 nm, 表明Ni(PO3)2-Ni2P对可见光有更宽的吸收范围. 其中, Ni(PO3)2-Ni2P-1表现出更宽的吸收带边, 这与文献[40]报道结果一致, Ni(PO3)2对可见光有更好的吸收作用. CdS NPs分别与Ni(PO3)2-Ni2P-1和Ni(PO3)2-Ni2P-2复合以后, 8%Ni(PO3)2-Ni2P-1/CdS NPs和8%Ni(PO3)2-Ni2P-2/CdS NPs的吸收带边分别红移至683和652 nm, 拥有更宽的光吸收范围, 吸收带边明显高于CdS NPs和Ni(PO3)2-Ni2P, 说明二者产生了良好的结构构建; Ni(PO3)2-Ni2P-2的自身带边相对较低, 但是CdS NPs与其复合后的带边提升幅度更大, 说明高含量的Ni2P更有利于异质结的构建. Ni(PO3)2的高吸光性和Ni2P促进异质结构建的性能, 两者的协同作用提高了Ni(PO3)2-Ni2P/CdS NPs复合催化剂的光吸收范围. 并且, 通过Tauc plot方法计算了半导体的禁带宽度, 由图5(B)可见, CdS NPs, Ni(PO3)2-Ni2P-1和Ni(PO3)2-Ni2P-2的带隙(g)分别为2.40, 2.06和2.34 eV, 超声复合后带隙分别降低至1.86 eV[8%Ni(PO3)2-Ni2P-1/CdS NPs]和2.13 eV[8%Ni(PO3)2-Ni2P-2/CdS NPs], 降低规律符合吸收带边的变化规律, 并且可以看出, 8%Ni(PO3)2-Ni2P-1/CdS NPs具有更适宜的可见光吸收带隙.
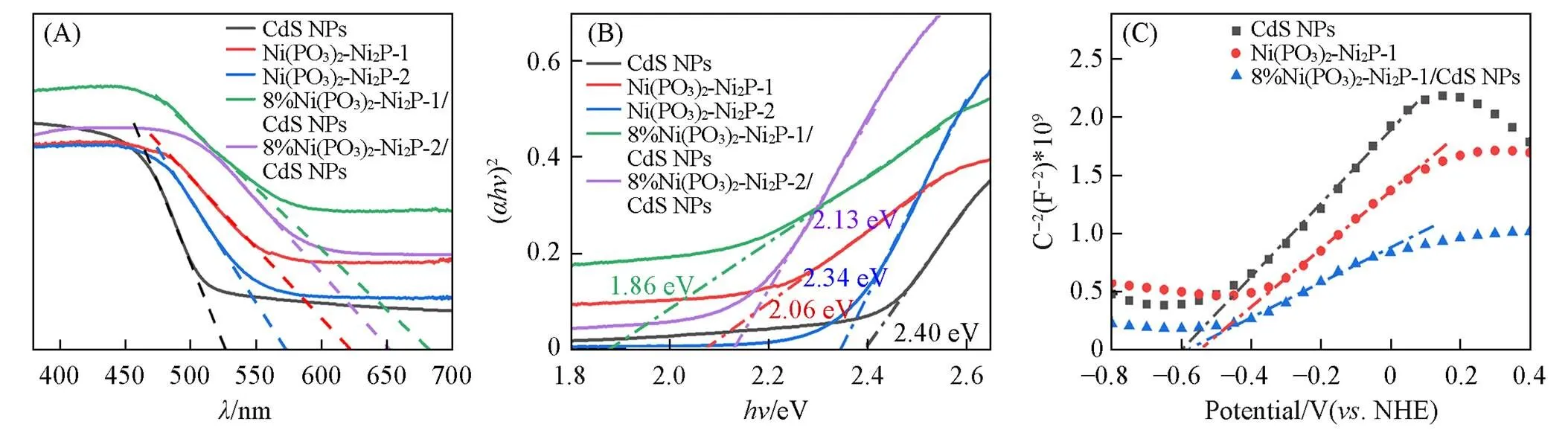
Fig.5 UV⁃Vis absorption spectra(A) and band gap(B) of CdS NPs, Ni(PO3)2⁃Ni2P⁃1, Ni(PO3)2⁃Ni2P⁃2, 8%Ni(PO3)2⁃Ni2P⁃1/CdS NPs and 8%Ni(PO3)2⁃Ni2P⁃2/CdS NPs, and Mott⁃Schottky plots(C) of CdS NPs, Ni(PO3)2⁃Ni2P⁃1 and 8%Ni(PO3)2⁃Ni2P⁃1/CdS NPs
用Mott-Schottky方法估算了CdS NPs, Ni(PO3)2-Ni2P-1和8%Ni(PO3)2-Ni2P-1/CdS NPs的导带 电势(CB)[55], 进一步阐明了光催化机理[图5(C)]. CdS NPs和Ni(PO3)2-Ni2P-1的CB分别为‒0.60和 ‒0.54 eV(. NHE, pH=0). 结合g, 通过价带电势公式(VB=g+CB)得到, CdS NPs和Ni(PO3)2-Ni2P-1的VB分别为1.80和1.52 eV. 这说明电子可以轻易从CdS NPs转移到Ni(PO3)2-Ni2P. 表1列出了相关数据.

Table 1 Band gap(Eg), conduction band(ECB) and valence band(EVB) of CdS NPs, Ni(PO3)2⁃Ni2P⁃1 and 8%Ni(PO3)2-Ni2P-1/CdS NPs
2.2 光催化材料的产氢性能
图6(A)中给出了CdS NPs, Ni(PO3)2-Ni2P-1, Ni(PO3)2-Ni2P-2, 8%Ni(PO3)2-Ni2P-1/CdS NPs和 8%Ni(PO3)2-Ni2P-2/CdS NPs光催化分解水产氢的活性. 未负载Ni(PO3)2-Ni2P时, CdS NPs的光催化活性(217 μmol·g‒1·h‒1)较低, 而Ni(PO3)2-Ni2P自身未表现出产氢催化活性. 当少量Ni(PO3)2-Ni2P-1(1.0%)与CdS NPs复合后, 产氢速率明显提高, 说明Ni(PO3)2-Ni2P-1是一种有效的助催化剂, 促进了光生载流子的分离和转移, 加速了质子还原. 随着Ni(PO3)2-Ni2P-1负载量的增加, Ni(PO3)2-Ni2P-1/CdS NPs光催化产氢活性呈先升后降的趋势, 负载量为1%~10%的Ni(PO3)2-Ni2P-1/CdS NPs光催化剂的产氢活性分别为负载前的10.1倍、 14.2倍、 15.5倍、 19.5倍和12.1倍. 当Ni(PO3)2-Ni2P-1负载量为8%时, 最高产氢活性可达到4237 μmol·g‒1·h‒1, 继续增加负载量, 光催化析氢速率降低, 可能是因为 Ni(PO3)2-Ni2P本身没有光催化性能, 主要是促进光吸收以及光生电荷分离, 而对光催化起主导作用的是CdS NPs, 应保证CdS NPs的适宜质量比. 同样是8%的 Ni(PO3)2-Ni2P-2/CdS NPs产氢活性是2982 μmol·g‒1·h‒1, 远低于8%Ni(PO3)2-Ni2P-1/CdS NPs的产氢活性, 说明Ni(PO3)2和Ni2P两者的比例对产氢活性具有重要影响. 推测原因: (1) Ni(PO3)2-Ni2P-1/CdS NPs 的吸收边波长更大, 带隙更小, 对可见光吸收范围更广, 并且Ni(PO3)2中官能团之间存在静电相互作用和氢键等弱相互作用[39], 更有利于电荷分离, 提高了光催化剂的电流密度, 达到增强光催化活性的目的; (2) 具有较高面密度的Ni(PO3)2可以有效协同活性成分Ni2P, 使其能够创造出更多的活性中心, 从而提高光催化剂的反应性和稳定性; (3) Ni(PO3)2-Ni2P-2以纳米颗粒的状态与CdS NPs复合重整, 颗粒堆积包覆可能会影响CdS NPs的光吸收, 没有CdS NPs沉积在Ni(PO3)2-Ni2P-1片层上的光吸收性能好, 从而影响产氢性能.
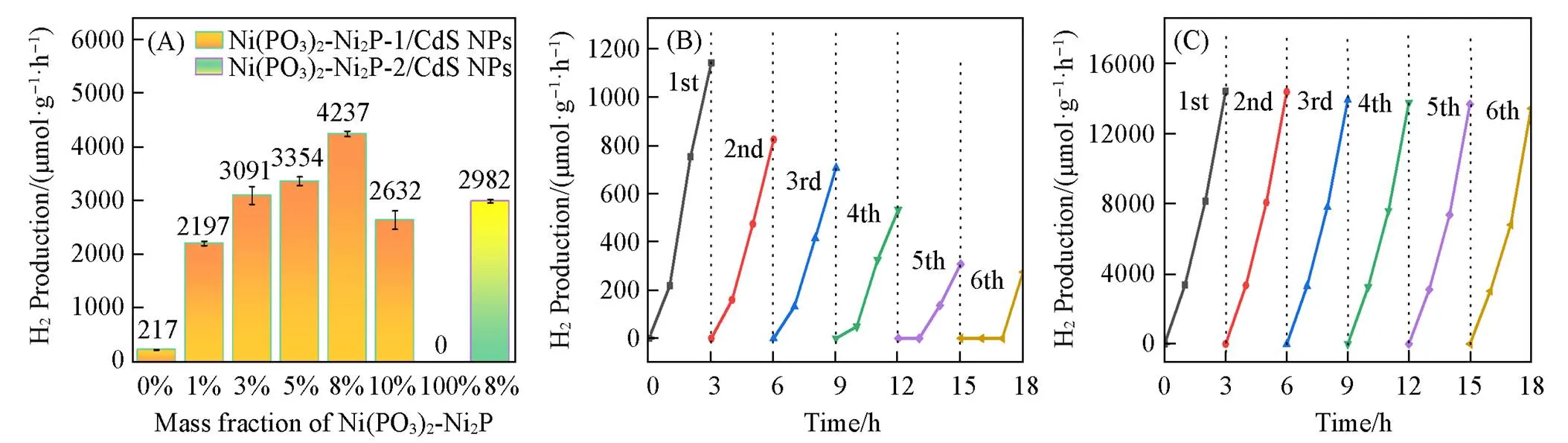
Fig.6 Photocatalytic H2 generation rates(A) of samples, time⁃circle experiment of photocatalytic H2 generation over the CdS NPs(B) and 8%Ni(PO3)2⁃Ni2P⁃1/CdS NPs(C)
光催化剂的耐久性和稳定性是其具有实际应用价值的重要方面[56]. 图6(B)和(C)分别给出了CdS NPs 和8%Ni(PO3)2-Ni2P-1/CdS NPs产氢循环的实验结果, 用以揭示其光催化稳定性. CdS NPs产氢活性随循环次数的增加而迅速下降, 在光催化循环10 h后, 产氢速率下降至初始值的47%. 而Ni(PO3)2-Ni2P-1/CdS NPs在18 h的循环实验中, 光催化产氢速率并没有明显减少, 反应进行到第6次循环时, 产氢速率仍可达到初始的89%, 表明Ni(PO3)2-Ni2P-1/CdS NPs复合材料具有良好的光催化稳定性, 在体系中完全没有贵金属的情况下获得了良好的催化性能.
光生电子-空穴对的分离和快速迁移是光催化产氢的关键步骤, 可以用稳态光致发光光谱和交流阻抗谱来表征[57]. 荧光发射光谱来源于光致电荷复合, 能极好地反映光生电子和空穴的分离效率[58]. 通常, 较高的光激发电子和空穴复合效率会导致较低的光催化活性[59]. 图7给出了CdS NPs, Ni(PO3)2-Ni2P-1, Ni(PO3)2-Ni2P-2, Ni(PO3)2-Ni2P-1/CdS NPs和Ni(PO3)2-Ni2P-2/CdS NPs的荧光发射光谱, 激发波长为340 nm. 可见, Ni(PO3)2-Ni2P-1表现出较强的荧光强度, 而Ni(PO3)2-Ni2P-2表现出了更强的荧光强度, 说明助催化剂容易发生光生载流子复合; Ni(PO3)2-Ni2P-1的荧光强度相对低一些, 说明Ni(PO3)2含量高可减轻光生载流子的复合. CdS NPs也表现出较强的荧光强度, 反映了光激载流子的高复合率, 从而导致低的光催化活性. CdS NPs分别与Ni(PO3)2-Ni2P-1和Ni(PO3)2-Ni2P-2复合后, 复合催化剂在525 nm处的荧光强度均下降最为明显, 说明CdS NPs与Ni(PO3)2-Ni2P已经产生异质结构, 能够将CdS NPs上产生的电子优先转移到Ni(PO3)2-Ni2P共催化剂上, 从而降低了电子-空穴对重组的速率. CdS NPs与Ni(PO3)2-Ni2P之间电子的快速转移导致荧光发生猝灭, 载流子分离能力和迁移效率极大提升, 从而具有最佳的光催化产氢活性.
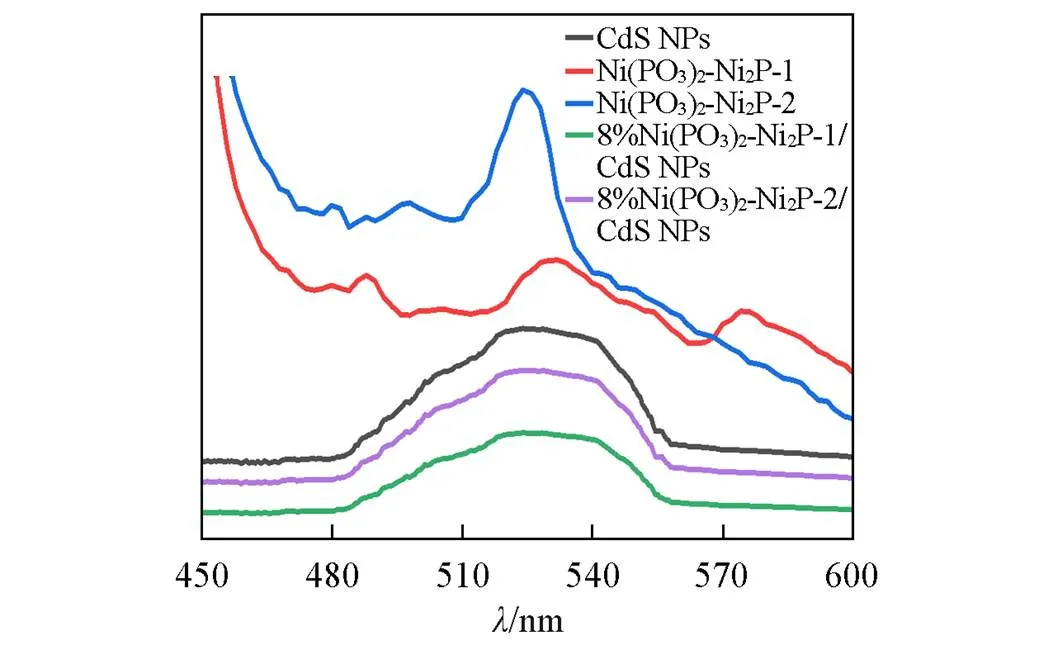
Fig.7 PL spectra of samples
图8为CdS NPs, Ni(PO3)2-Ni2P-1, Ni(PO3)2-Ni2P-2, 8%Ni(PO3)2-Ni2P-1/CdS NPs以及8%Ni(PO3)2-Ni2P-2/CdS NPs的电化学阻抗(EIS)谱图和瞬态光电流响应(-)谱图, 进一步证明CdS NPs与Ni(PO3)2-Ni2P复合后在光生电荷转移过程中存在的优势[60]. 如图8(A)所示, CdS NPs的半圆半径最大, 表现出较高的阻抗, 分离能力弱; Ni(PO3)2-Ni2P-2的半圆半径明显高于Ni(PO3)2-Ni2P-1, 表明Ni(PO3)2含量高更有利于光生电子的分离; 8%Ni(PO3)2-Ni2P-1/CdS NPs和8%Ni(PO3)2-Ni2P-2/CdS NPs均有较小的半圆半径, 说明电荷转移阻力小, 都有光生电子与空穴快速分离的能力; 并且, CdS NPs与Ni(PO3)2-Ni2P-2复合后, 半圆半径变化幅度最大, 可能是Ni(PO3)2-Ni2P-2中Ni2P的含量高, 更有利于异质结的构建. 由图8(B)可见, CdS NPs, Ni(PO3)2-Ni2P-2, Ni(PO3)2-Ni2P-1, 8%Ni(PO3)2-Ni2P-2/CdS NPs以及 8%Ni(PO3)2-Ni2P-1/CdS NPs的光电流密度依次增大, 复合后催化剂均有较高的光电流密度, 说明 Ni(PO3)2-Ni2P的引入有效促进了CdS NPs光生载流子的转移, 从而提高了复合材料的产氢活性. 并且可看出, Ni(PO3)2-Ni2P-1/CdS NPs的性能更优, Ni(PO3)2含量高更有利于光生载流子的转移. 当关闭光源时, 光电流密度降为零, 表明在模拟可见光照射下光生电子只产生并转移到氧化铟锡(ITO)电极上.

Fig.8 EIS Nyquist plots(A) and transient photocurrent response spectra(B) of samples
2.3 光催化机理
通常, 太阳能驱动的半导体光催化活性主要依赖于其较强的光吸收能力、 高效的电荷分离和快速迁移特性, 以及适宜表面进行氧化还原反应的半导体性质. 基于上述结果, 对Ni(PO3)2-Ni2P/CdS NPs复合光催化材料提出了可能的电荷转移和光催化机理(Scheme 2).
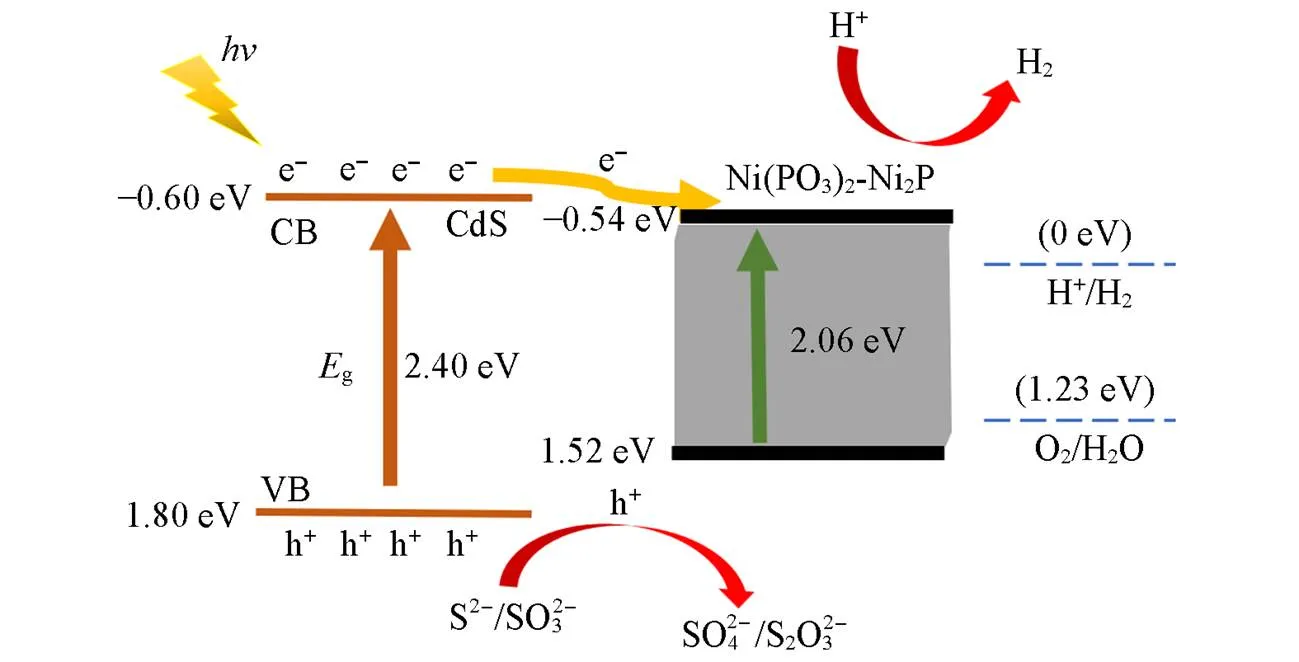
Scheme 2Mechanism of photocatalytic reaction for Ni(PO3)2⁃Ni2P/CdS NPs to hydrogen
首先, 在可见光照射下, CdS NPs价带上的电子会被激发跃迁到导带上, 形成电子-空穴对. Ni(PO3)2-Ni2P/CdS NPs增强的可见光吸收特性有助于产生更多的电子和空穴, 有利于光催化活性的提高. 然后, 设计的异质结极大地促进了电子和空穴的分离. 得益于CdS NPs和Ni(PO3)2层片阵列之间适宜的能带结构和界面作用, Ni(PO3)2-Ni2P作为电子陷阱捕获了CdS NPs中的光生电子, CdS NPs的光生电子通过界面转移到Ni(PO3)2-Ni2P上, 从而有效地实现电子-空穴对的分离. 因其较低的费米能级, 具有更强的电子捕获能力, 能够使光生电子快速迁移, 从而极大地降低了CdS NPs的光生电子-空穴对的复合率. 最后, Ni(PO3)2-Ni2P的引入可以降低H+的还原过电位, 并作为催化活性位点. Ni(PO3)2-Ni2P的导带低于CdS NPs, 有利于H2的产出. 根据文献[61]的报道, 将光生电子注入到磷化物和磷酸盐中, 可以为H2的生产提供更有效的能量, 为电子在光催化剂中的有效转移提供了保证. 此外, 在Ni2P共催化剂中积累了部分电子, 以协助H+还原为H2, 从而促进复合光催化剂表面更好地进行H2生成反应. Ni(PO3)2-Ni2P/CdS NPs产生的空穴被Na2S和Na2SO3的混合溶液所消耗, 复合材料的光催化产氢活性得到大幅提高.
3 结 论
采用共催化剂改性与异质结构建策略, 以花球状Ni(OH)2为前驱体, 利用热磷酸化技术, 并结合简便的超声法, 合成了一种比例可调、 高效、 稳定、 廉价的非贵金属可见光催化分解制氢催化材料 Ni(PO3)2-Ni2P/CdS NPs.当Ni(PO3)2-Ni2P-1负载量为8%时, 该光催化体系的产氢速率最高, 达到4237 μmol·g‒1·h‒1, 约为CdS NPs产氢速率的19倍. Ni(PO3)2-Ni2P修饰CdS NPs的协同效应, 明显增强了可见光吸收特性、 有效促进了光生电子-空穴对的分离和快速迁移, 作为催化活性位点降低了H+的还原过电位, 从而极大地提高了CdS NPs的光催化产氢活性. 在共催化剂Ni(PO3)2-Ni2P中, Ni(PO3)2和Ni2P可协同作用促进光吸收以及光生电荷分离, Ni(PO3)2含量的增加更有利于促进光吸收和光生电荷分离, Ni2P更有利于异质结的构建. 所制得的复合光催化材料在光催化循环产氢实验中也表现出很好的稳定性. 为可见光驱动光催化剂分解水产氢在解决能源和环境问题上提供了更多的可能.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20220050.
[1] Zhang Y. Z., Zhou W., Tang Y., Guo Y. C., Geng Z. K., Liu L. Q., Tan X., Wang H. Y., Yu T., Ye J. H.,, 2022,, 121055
[2] Li Y. X., Zhang W. Z., Li H., Yang T. Y., Peng S. Q., Kao C., Zhang W. Y.,, 2020,, 124928
[3] Chang J. F., Lv Q., Li G. Q., Ge J. J., Liu C. P., Wei X.,, 2017,, 486—496
[4] Liu Y. Y., Li B. J., Xiang Z. H.,, 2021,(34), 2007576
[5] Fan H. T., Wu Z., Liu K. C., Liu W. S.,, 2022,, 134474
[6] Liu H. W., Chen J., Guo W. Y., Xu Q. J., Min Y. L.,, 2022,, 652—660
[7] Nuray G.,, 2020,, 146442
[8] Wang Y. Q., Xu X. X., Lu W., Huo Y. Q., Bian L.,, 2018,(12), 4219—4227
[9] Liu Q. Y., Qi Y. L., Zheng Y. F., Song X. C.,, 2017,(7), 1329—1333
[10] Tonda S., Kumar S., Gawli Y., Bhardwaj M., Ogale S.,, 2017,(9), 5971—5984
[11] Jo W. K., Selvam N. C. S.,, 2017,, 913—924
[12] Liu C., Xiong M. H., Chai B., Yan J. T., Fan G. Z., Song G. S.,, 2019,(24), 6929—6937
[13] Jian Q., Hao X. Q., Jin Z. L., Ma Q. X.,, 2020,(4), 1932—1943
[14] Qiao S. S., Feng C., Guo Y., Chen T. X., Akram N., Zhang Y., Wan W., Yue F., Wang J. D.,, 2020,, 125068
[15] Xin X., Song Y., R. Guo S. H., Zhang Y. Z., Wang B. L., Wang Y. J., Li X. H.,, 2020,, 154635
[16] Reddy D. A., Park H., Ma R., Kumar D. P., Lim M., Kim T. K.,, 2017,(7), 1563—7150
[17] Han B., Liu S., Zhang N., Xu Y. J., Tang Z. R.,, 2017,, 298—304
[18] Xie Y. P., Yu Z. B., Liu G., Ma X. L., Cheng H. M.,, 2014,(6), 1895—1961
[19] Cao W. Y., Zhang X. L., Zheng Y. J., Wang K., Dai H. T.,, 2017,(5), 2924—2930
[20] Li Y. K., Zhang P., Wan D. Y., Xue C., Zhao J. T., Shao G. S.,, 2020,, 144361
[21] Meng X. J., Kuang W. D., Qi W. L., Cheng Z. X., Thomas T. J., Liu S. Q., Yang C., Yang M. H.,, 2020,(10), 2752—2759
[22] Huo J. P., Lei Y., Zeng G., Zeng H.,, 2014,(29), 11040—11044
[23] Wei L., Adamson M. A. S., Vela J.,, 2020,(8), 1179—1185
[24] Bown J. J., Page A. J.,, 2019,(21), 13029—13035
[25] Gao N., Zhou Y. K., Shen S. B.,, 2019,(12), 5372—5379(高宁, 周玉康, 沈树宝. 化工进展, 2019,(12), 5372—5379)
[26] Osterloh F. E.,, 2007,(1), 35—54
[27] Gultom N. S., Abdullah H., Kuo D. H.,, 2020,, 118985
[28] Xu Z. H., Zhu Q. H., Xi X. G., Xing M. Y., Zhang J. L.,, 2021,(3), 678—686
[29] Jiao Y. Y., Li Y. K., Wang J. S., He Z. H., Li Z. J.,, 2020,, 147603
[30] Yang H., Yang C., Zhang N. N., Mo K. L., Li Q., Lv K. L., Fan J. J., Wen L. L.,, 2021,, 119801
[31] Fang Y. Z., Liu R. Z., Dou S. X., Shang Q. Q., Wang D. T., Kong X. J., Liu J. H.,, 2022,(12), 7724—7737
[32] Luo M. H., Yao W. F., Huang C. P., Wu Q., Xu Q. J.,, 2015,(26), 13884—13891
[33] Dong Q. W., Li M. L., Sun M. S., Si F. Y., Gao Q. Z., Cai X., Xu Y. H., Yuan T., Zhang S. S., Peng F., Fang Y. P., Yang S. Y.,, 2021,(11), 2100878
[34] Shen R. C., Xie J., Xiang Q. J., Chen X. B., Jiang J. Z., Li X.,, 2019,(3), 240—288(沈荣晨, 谢君, 向全军, 陈小波, 江吉周, 李鑫. 催化学报, 2019,(3), 240—288)
[35] Yu Y. H., Chen Q. R., Li J., Rao P., Li R. S., Du Y. L., Jia C. M., Huang W., Luo J. M., Deng P. L., Shen Y. J., Tian X. L.,, 2022,, 1091—1102
[36] Wu T. F., Wang P. F., Qian J., Ao Y. H., Wang C., Hou J., Fan G. Z., Song G. S.,, 2017,(40), 13793—13801
[37] Zhang C., Chu S. P., Liu B. Q., Liu Y., Guo Z. M., Lv Z. G.,, 2021,, 150987
[38] Li K., Ma J. W., Guan X. L., He H. W., Wang M., Zhang G. L., Zhang F. B., Fan X. B., Peng W. C., Li Y.,, 2018,(47), 22173—22179
[39] Tong J. H., Li W. Y., Bo L. L., Li Y. L., Li T., Zhang Q.,, 2019,, 134579
[40] Ray A., Sultana S., Paramanik L., Parida K. M.,, 2020,(37), 19196—19245
[41] Samal A., Swain S., Manju U., Das D. P.,., 2019,(11), 10052—10063
[42] Jiang H., Zhao T. Y., Yang Z. K., Xing Z. P., Li Z. Z., Zou J. L., Pan K., Zhou W.,, 2019,, 160—167
[43] Gurbani N., Choudhary R. J., Phase D. M., Marumoto K., Liu R. S., Chouhan N.,, 2021,, 100105
[44] Wang Y., Pan Q., Jia K., Wang H. B., Gao J. J., Xu C. L., Zhong Y. J., Alshehri A. A., Alzahrani K. A., Guo X. D., Sun X. P.,, 2019,(10), 6579—6583
[45] Li Y. W., Xu W. Q., Xie Z. P., Zhang L. Z., Yao J. H.,, 2017,(7), 1625—1636
[46] Liu Z. L., Li B. Q., Feng Y. J., Jia D. C., Li C. C., Sun Q. F., Zhou Y.,, 2022,, 151384
[47] Norouzi P., Karimpour A., Ganjali M. R.,, 2019,(17), 16184—16194
[48] Sheng Q., Li X., Prins R.,, 2021,(20), 11180—11183
[49] Irfan R. M., Tahir M. H., Khan S. A.,, 2019,, 1—9
[50] Yan C. Y., Li W. Q., Liu X. J., Chen M., Liu X., Li X. M., Zai J. T., Qian X. F.,, 2021,, 48872—48880
[51] He K., Tsega T. T., Liu X., Zai J.T., Li X. H., Liu X. J., Li W. H., Ali N., Qian X. F.,, 2019,(34), 11903—11909
[52] Yang X., Lu A. Y., Zhu Y.,, 2015,, 634—341
[53] Sun Z., Zheng H., Li J.,, 2015,(9), 2668—2676
[54] Ran J., Zhang J., Yu J.,, 2014,(22), 7787—7812
[55] Pan Y. X., Peng J. B., Xin S.,, 2017,(6), 5449—5456
[56] Li X., Yu J., Low J.,, 2015,(6), 2485—2534
[57] Xiang Q., Cheng F., Lang D.,, 2016,(9), 996—1002
[58] Li Y., Wang L., Cai T.,, 2017,, 366—374
[59] Gong H. S., Li Z., Chen Z. H., Liu Q. W., Song M. X., Huang C. J.,, 2020,(4), 3665—3674
[60] Yuan J. L., Wen J. Q., Zhong Y. M., Li X., Fang Y. P., Zhang S. S., Liu W.,, 2015,(35), 18244—18255
[61] Cheng X. D., Pan Z. Y., Lei C. J., Jin Y. J., Yang B., Li Z. J., Zhang X. W., Lei L. C., Yuan C., Hou Y.,, 2019,(3), 965—971
Heterostructure Construction of Noble-metal-free Ternary Composite Ni(PO3)2-Ni2P/CdS NPs and Its Visible Light Efficient Catalytic Hydrogen Production
WANGGuangqi1, BIYiyang2, WANGJiabo2, SHIHongfei2, LIUQun2*, ZHANGYu2*
(,,,132022,)
The development of visible-light-driven efficient, stable, and inexpensive photocatalysts for water- splitting hydrogen production is an important topic to address global energy and environmental challenges. In this work, a highly efficient, stable, and non-noble metal CdS-based photocatalytic composite Ni(PO3)2-Ni2P/CdS NPs was synthesized by combining cocatalyst modification and heterostructure construction strategy. Binary cocatalyst Ni(PO3)2-Ni2P nanosheet arrays were prepared using flower bulb Ni(OH)2as precursor by gas-phase sintering phosphating process, and CdS NPs were combined with them to form a new type of ternary composite photocatalyst [Ni(PO3)2-Ni2P/CdS NPs] by sonochemical. The photocatalytic hydrogen production rate of 8%(mass fraction) Ni(PO3)2-Ni2P/CdS NPs reaches 4237 μmol·g‒1·h‒1with Na2S-Na2SO3as sacrificial agent under the irradiation of visible light(>420 nm), which is about 19 times for CdS NPs(217 μmol·g‒1·h‒1). In the cycling experiment, the hydrogen production rate of reaction is about 89% of the initial value after sixth cycles(18 h), which demonstrates excellent catalytic stability. Compared with CdS NPs, the absorption edge of Ni(PO3)2-Ni2P/CdS NPs is obviously red-shifted, the forbidden band width is reduced to 1.86 eV, and the overpotential of H+reduction is reduced, which exhibits strong light absorption properties and a suitable band gap structure. The photogenerated electrons of CdS NPs are transferred to Ni(PO3)2-Ni2P through the synergistic effect between Ni(PO3)2-Ni2P and CdS NPs, which effectively promotes the separation of photogenerated carriers and improves the activity and stability of hydrogen production. This noble-metal-free composite photocatalyst may provide a way to design very efficient water splitting catalysts, which will promotes the practical industrial application of CdS visible photocatalytic water splitting.
CdS NPs; Ni(PO3)2; Ni2P; Photocatalysis; Hydrogen evolution
O643.3
A
10.7503/cjcu20220050
2022-01-21
2022-04-09.
张 钰, 女, 博士, 教授, 主要从事催化化学研究. E-mail: zhang99yu@jlict.edu.cn 刘 群, 男, 博士, 讲师, 主要从事材料化学研究. E-mail: QunLiu@jlict.edu.cn
吉林省科技发展计划项目(批准号: 20190103117JH)资助.
Supported by the Science and Technology Development Program of Jilin Province, China(No.20190103117JH).
(Ed.: Y, K, S)
猜你喜欢
物理化学学报(2024年11期)2024-11-06 00:00:00
——潘桂棠光生的地质情怀
沉积与特提斯地质(2021年2期)2021-07-20 06:33:26
无机盐工业(2017年5期)2017-05-25 00:37:34
物理化学学报(2017年3期)2017-03-11 00:25:30
化工管理(2017年25期)2017-03-05 23:32:36
功能材料(2016年1期)2016-05-17 03:38:24
材料科学与工程学报(2016年5期)2016-02-27 07:11:28
昭通学院学报(2016年5期)2016-02-24 10:51:12
石家庄铁道大学学报(自然科学版)(2015年3期)2015-02-28 15:05:43
影像科学与光化学(2014年5期)2014-03-11 16:03:22
