
COL1A2基因剪接突变所致成骨不全1例分析
2022-07-13 11:34王天平胡曼云贵州省贵阳市第四人民医院内分泌科550005
医学理论与实践 2022年13期
王天平 胡曼云 张 喜 贵州省贵阳市第四人民医院内分泌科 550005
成骨不全症(Osteogenesis imperfecta,OI)或称为脆骨病,是常见的单基因突变所致的遗传性骨病,遗传方式呈常染色体显性遗传(Autosomal dominant,AD)和常染色体隐性遗传(Autosomal recessive,AR),以AD多见,但也有不少散发病例的报道。临床上以骨量低下、骨骼脆性增加而反复发生骨折、骨骼畸形为主要特征,可伴有蓝巩膜、牙本质发育不良、听力障碍、皮肤及韧带松弛等骨骼外表现。主要涉及的致病基因是骨基质Ⅰ型胶原蛋白编码基因及其代谢相关基因[1-2]。
1 病例资料
患儿男性,12岁,汉族,11岁时因爬楼踩滑(约于站立高度)致右侧肩部及右膝关节骨折,外院行手术治疗,随后逐渐出现腰背部持续性隐痛,疼痛能耐受,行走时加重,日常活动(慢走、短距离爬楼)无明显受限,但跑、跳等运动完成困难,上述症状持续约1年后再次就诊外院,查维生素D:25-羟基维生素D2+D316.8ng/ml。腰椎片:椎体骨骺板骨软骨炎可能。遂转诊于我院骨科门诊查胸、腰椎MRI(见图1):脊柱稍向后凸,胸腰段椎体变扁及信号增高原因,性质:骨质疏松可能。随后以“成骨不全?”收入院。入院后详细询问病史,患儿出生时巩膜就为蓝色,约1岁时开始口服“鱼肝油等”药物,坚持饮牛奶200~400ml/d,持续2~3年后上述行为调整为间断进行。患儿日常活动能力与其他同龄儿无明显差别。患儿饮食上喜素食,近2年体重增加10+kg,身高增高约3cm。此次主要因为腰背部持续性疼痛就诊欲查明原因。
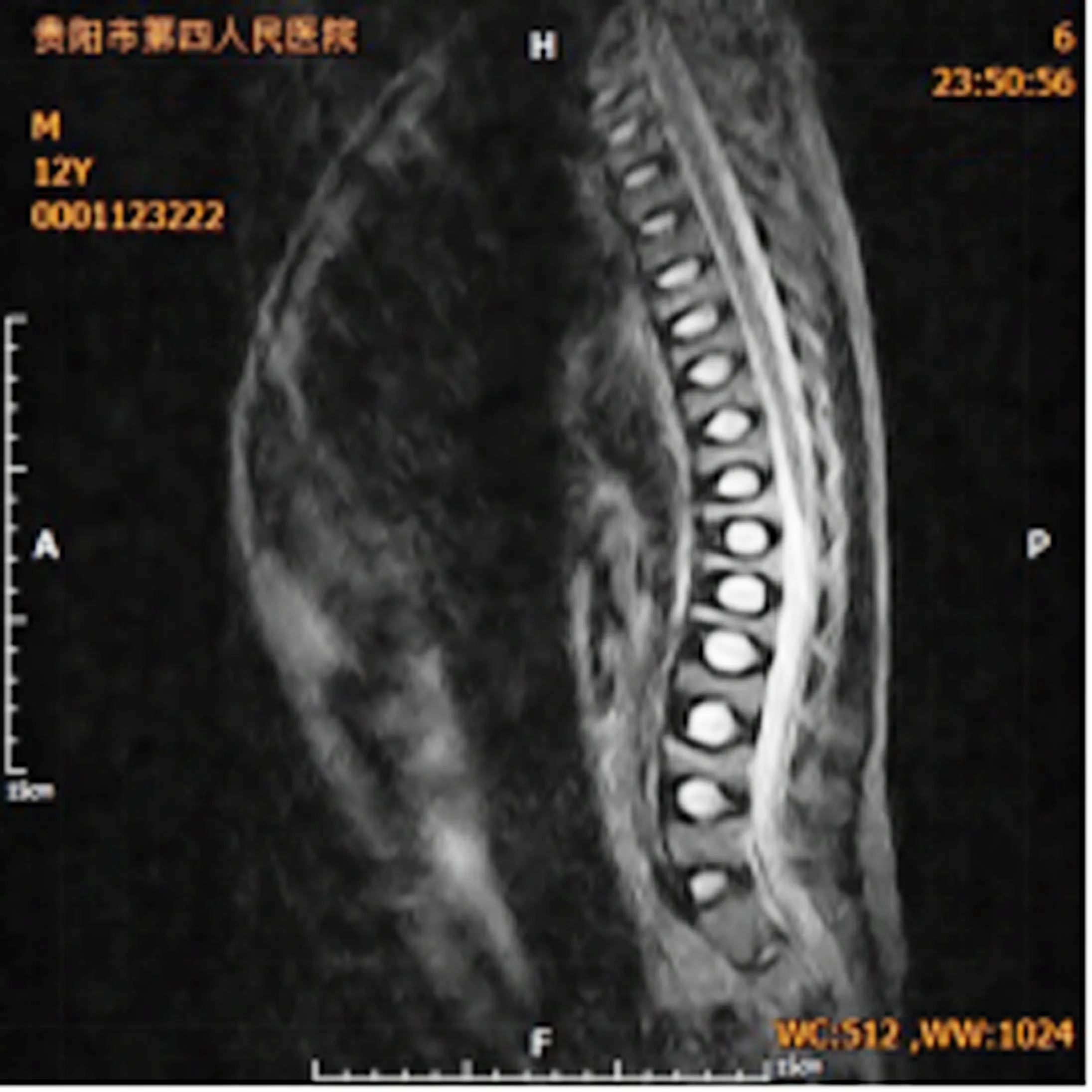
图1 腰椎MRI
患儿母亲孕期未产检,患儿囟门5~6岁才完全闭合,时间较同龄儿明显滞后,其他生长发育方面患儿父母未发现明显异常。目前还未出现遗精等青春期启动表现。9岁时因“左侧小脑少突星形角质细胞瘤并出血性卒中”于外院手术治疗。患儿直系亲属三代中均无身高异常矮、骨骼明显畸形及脆性骨折情况。
体格检查:生命体征平稳,身高:133cm,体重:35kg,指间距:137cm,上部量:73cm,下部量:60cm,坐高:70cm。双眼巩膜呈蓝色。牙列稀疏。双侧腹股沟触诊未扪及包块。双侧髂骨于腹部中部外侧可扪及。脊柱胸腰段后凸畸形。双手指关节弯曲畸形(见图2),未见多指、趾,四肢指、趾关节均较柔软(见图3)。双下肢稍呈“X”形表现,行走稍异常。余查体未见明显异常。

图2 双手正面照片
辅助检查及分析:(1)骨代谢相关的指标:PINP>1 200.00ng/ml,CTX 0.897ng/ml,甲状旁腺激素 42.20pg/ml,骨源性碱性磷酸酶126.85μg/L,其中反映骨生成的指标PINP明显增高,考虑为骨折后骨生成明显增加有关。(2)胸腰椎正侧位+骨盆+双手正位片:①符合成骨不全X线征象;②双手未见明显异常。阅片可见骨质稀疏,骨小梁稀少。(3)染色体检查提示46 XY,正常男性染色体表现。性激素未见异常。阴囊B超提示双侧睾丸存在。结合腹股沟查体情况不考虑性功能不全所致的继发性骨质疏松。(4)皮质醇昼夜节律试验提示皮质醇呈昼夜节律变化。结合查体情况不考虑皮质醇增多症所致的继发性骨质疏松。(5)五官科会诊诊断考虑右耳传导性聋(轻度)、脆骨—蓝巩膜—耳聋综合征。其他辅查未见明显异常。
治疗:本例患儿前来我院就诊时已经出现了右侧肩部、右膝关节骨折及胸腰椎的多发压缩性骨折,生活方式指导患儿提高肌肉强度及改善机体协调能力,注意避免频繁跌倒所致骨折发生,并进行补钙及补充维生素D3,因患儿家属拒绝,住院期间没有使用双磷酸盐抑制骨吸收治疗,在之后的随访中,患儿再次发生了下肢的骨折,就诊于外院手术治疗骨折后,使用了双磷酸盐药物抑制骨吸收。建议患儿的父母在下次备孕前进行有效的遗传咨询和怀孕后产前诊断,这有利于OI家庭的优生优育。
2 基因检查及家系验证
基因检查结果:患儿存在COL1A2基因c.693+1G>C剪接突变(Het)。使用Sanger测序对家系成员验证该突变位点,结果提示未见其他家系成员存在相同突变。Sanger测序图谱见图4。
患儿存在COL1A2基因c.693+1G>C剪接突变,为杂合突变;患儿父亲、患儿母亲、患儿大妹、患儿二妹测序结果未见相同突变
3 讨论
患儿为1例携带COL1A2基因c.693+1G>C剪接突变(Het)的散发OI病例。根据患儿疾病的严重程度及临床特征,考虑患儿OI Ⅰ型(Sillence分型)。患儿突出的临床表现是反复发生脆性骨折,伴有蓝巩膜、听力障碍、皮肤及韧带松弛体征,基因检查示COL1A2基因c.693+1G>C剪接突变(Het),Sanger测序行家系验证未见其他家系成员存在相同突变。赵秀丽等[3]已通过研究构建了中国人OI相关的COL1A1/2基因突变谱,本例患者存在的COL1A2基因c.693+1G>C剪接突变在其突变谱中是可以查询到的,证实该突变在中国人群中是OI的致病突变。
Ⅰ型胶原蛋白是骨有机质中含量最丰富的蛋白质,其代谢异常是OI发病的重要环节。Ⅰ型胶原蛋白分子由2条α1链和1条α2链构成稳定的异源三螺旋结构,α1链和α2链分别由COL1A1和COL1A2基因编码,若COL1A1或COL1A2基因发生突变,其翻译的前α1链或前α2链将形成异常三螺旋结构的前Ⅰ型胶原蛋白,从而导致Ⅰ型胶原蛋白合成的数量减少或蛋白功能、结构异常[4]。导致OI的致病基因有很多,其中以COL1A1、COL1A2两种基因突变最多见,文献报道占80%~90%[5],而携带COL1A1/2基因致病突变的OI患者中,COL1A1基因突变的检出率会更高,目前报道的COL1A1基因突变有2 077种,COL1A2基因突变有1 089种(https://oi.gene.le.ac.uk,数据最后更新时间分别为2020年11月18日、2020年11月19日)。携带COL1A1/2基因突变的OI患者中,COL1A1基因突变的患者病情也会更严重,这与COL1A1、COL1A2基因分别编码的α1链、α2链在Ⅰ型胶原蛋白分子中的不同作用相关。COL1A1/2基因突变的类型有错义突变、剪接突变、插入和缺失、移码突变等,其中最常见的是错义突变[6],本例患儿存在的剪接突变并不是OI相关的COL1A1/2基因突变类型中最少见的,国内的两项研究,分别对56例OI患者[7]和200例OI先证者及其家系成员[3]进行COL1A1/2基因突变鉴定,剪接突变的占比分别为17.9%、15.2%,均大于缺失或移码突变等。
关于OI的疾病分型,1979年Sillence等将OI分为4型,主要根据患者的临床表现及病情严重程度来划分(传统分型):其中Ⅰ型最轻微,无肢体变形;Ⅱ型围产期死亡;Ⅲ型存在严重长骨畸形;Ⅳ型病情严重程度介于Ⅰ和Ⅲ之间,临床表现不一。国际骨骼人类遗传学疾病命名组织(INCDS)于2009年在Sillence分型的基础上增加了1型,将OI分为5型,主要分型依据也是根据临床表型和疾病严重度。这两种疾病分型方法是大多数文献中所提及的分型方法,这样的临床分型可以在不做基因检测的情况下迅速判断OI患者疾病的严重程度和临床表型特征。但OI涉及的致病基因很多,突变类型也多种多样,遗传外显性差异也较大,相同基因突变可能导致不同表现型,即使在同一家系也可能出现不同的表现型,目前国内外均已有针对很多疾病的基因靶向治疗,对于OI患者是否能进行这方面的治疗,有待更多学者的研究。那OI是否能根据致病基因对疾病进行分型以指导未来可能会有的基因靶向治疗呢?Fratzl-Zelman则在分子遗传学基础上,主要根据OI致病基因在疾病病理生理过程中的生化作用将OI分成了11型(Ⅰ~Ⅺ),还有部分致病基因未分型,其中Ⅰ~Ⅳ型的具体分型方法基本同传统分型方法。国内的学者[8]也提出了新的分型,根据患者的致病基因、遗传方式,将OI分成了15型,仍有部分致病基因未分型。可见,随着基因检测技术在临床上的广泛使用,以致病基因为主要的分型依据已有趋势。
COL1A2基因位于7q21.3,由52个外显子组成,COL1A2基因正常表达的第一步就是要以DNA为模板转录形成mRNA,初始转录产物形成的是mRNA前体,需要经过加工和修饰,才能形成有功能的mRNA,这个过程中有一个重要环节就是要在剪接酶的作用下将内含子去除,当基因突变出现在内含子并导致剪接位点发生了改变后,就会造成mRNA前体的异常剪接,进而编码出异常的前α2链,最终导致Ⅰ型胶原蛋白结构或功能异常。本例患儿携带COL1A2基因c.693+1G>C剪接突变(Het),该突变是发生在14号内含子,第693号核苷酸向后数1位的内含子核苷酸由G替换成了C,该突变就会导致mRNA前体的异常剪接,是已被报道的致病突变。进一步查询相关的突变数据库(https://databases.lovd.nl/shared/genes/COL1A2,访问时间2021年12月7日),c.693+1这个位点的突变已报道了5例,国内外均有报道,DNA改变均为碱基替换,疾病诊断均考虑OI Ⅰ型,说明该突变位点及DNA突变方式并不是中国人群特有的,该突变方式所致的疾病分型上中国人群并无明显异质性,当然这还需要更多数据的支持。
随着临床上基因检测技术的广泛使用,关于OI致病基因的文献报告层出不穷,的确丰富了OI致病基因的突变谱,但目前仍无针对OI致病基因的相关治疗,目前基本为对症治疗。
猜你喜欢
中南药学(2022年2期)2022-03-30
中国听力语言康复科学杂志(2021年6期)2021-12-21
消费者报道(2019年6期)2019-08-17
中国社区医师(2019年1期)2019-06-26
Coco薇(2017年12期)2018-01-03
中学生理科应试(2017年6期)2017-09-27
饮食与健康·下旬刊(2017年3期)2017-03-30
智能制造(2015年4期)2015-05-12
饮食科学(2009年12期)2009-12-11
