
硫酸钡粒径调控剂C-N-CDs的制备、应用及机理
2022-07-12 07:40胡广齐叶炜浩郭宝颜许晓凯胡超凡庄健乐刘应亮
无机化学学报 2022年7期
马 昱 胡广齐 叶炜浩 郭宝颜 许晓凯 胡超凡 庄健乐 刘应亮*,
(1华南农业大学材料与能源学院,广州 510642)(2岭南现代农业科学与技术广东省实验室,广州 510642)(3广东技术师范大学光电工程学院,广州 510665)
0 引 言
硫酸钡(BaSO4)是一种重要的重晶石资源,外观为白色粉末,分子式为BaSO4,相对分子质量为233.39,15℃下比重为4.50,熔点为1 580℃,具有较高的折射率(1.63~1.65);几乎不溶于水、稀酸、乙醇等[1]。BaSO4在涂料、塑料、纸张、橡胶、颜料油墨等领域得到广泛的应用,尤其是纳米BaSO4已经发展成为一种不可或缺的无机填充材料[2]。粒径小、粒径分布窄的纳米BaSO4颗粒具有优异的纳米增韧性能,可极大增强聚合物薄膜的韧性[3],因此制备粒径小、粒度分布窄的纳米BaSO4颗粒一直是研究者们的工作热点。常见的制备纳米BaSO4的方法包括配位沉淀法、微乳液法以及微反应器法[4]。配位沉淀法制备出的纳米BaSO4颗粒的粒径比后2种方法制备的纳米BaSO4颗粒大,但具有工艺简单、成本低、技术成熟的特点。利用配位沉淀法制备出粒径更小的纳米BaSO4颗粒是非常有必要的,但也是极具挑战性的。
在过去的十年中,光致发光的新型零维碳纳米材料——碳点(CDs)因制备简单、光稳定性高、毒性低等特点,在光电转换[5-6]、光催化[7-9]、生物成像[10]等光学领域引起研究者的极大关注。CDs是一种近球形、直径小于10 nm的碳纳米材料,通常由C、H、O、N等基本元素组成,其核心是以sp2杂化为主的石墨型结构以及部分无定型碳结构组成的碳核,表面富含如羟基、氨基、羧基、酰胺基等多种官能团[11]。正是CDs丰富的表面官能团以及独特的空间结构赋予CDs优异的理化性能,具有广阔的应用前景,CDs理化功能的研究也逐渐成为新的研究热点。Zhu等将CDs中的一种——石墨烯量子点(GQDs)运用到制备Ca(OH)2中,制备出一种粒径小(约80 nm)、粒径分布相对均匀、聚集较少的Ca(OH)2/GQDs纳米复合材料[12]。He等使用GQDs作为分散剂将商业化石墨烯粉末分散在水中,取得了具有良好稳定性的石墨烯水分散体[13]。Hao等通过热分解合成了一种羧基碳量子点(CCQDs),其对CaSO4和BaSO4具有优异的阻垢性能:0~80℃静态试验下,较少的CCQDs添加量便可以达到100%的阻垢效率[14]。另外,不同CDs的理化性能在锂离子纳滤膜[15]、膨润土改性[16]、纳米CaCO3药物载体[17]、纳米 CaCO3形貌调控[18]、除锈剂[19]等方面都取得较好的应用。以上这些工作主要围绕CDs的化学性能、物理性能、空间结构3个方面进行研究并加以应用。
本文中报道了CDs的一项创新性应用:利用其化学性能调控纳米BaSO4颗粒粒径。以柠檬酸与乙二胺为前驱体,通过水热反应制备出平均粒径约2 nm、表面富含羧基与氨基、带负电的CDs。不同于已报道的通过双亲性调控Ca(OH)2颗粒粒径的GQDs,研究发现C-N-CDs与传统配位剂乙二胺四乙酸(EDTA)类似,富含羧基与氨基的表面官能团赋予C-N-CDs优异的配位能力,可减缓结晶从而减小BaSO4颗粒的粒径。另外,C-N-CDs在反应过程中被镶嵌在晶核表面,其表面负电荷产生的静电斥力防止BaSO4晶核的聚集、微小的类球体空间结构造成的空间位阻阻碍BaSO4晶体的生长,最终制备出的BaSO4颗粒平均粒径为45.3 nm,小于传统配位剂EDTA调控下BaSO4颗粒的平均粒径73.7 nm。将制备的BaSO4样品按照8%的质量分数添加入聚乙烯醇(PVA)薄膜能够取得更为优异的纳米增韧效果。最后,C-N-CDs还具有制备简单、低毒、生物相容性好[20-21]等诸多优点,应用前景广阔。
1 实验部分
1.1 试剂
柠檬酸(分析纯)、乙二胺(纯度 99%)、EDTA(分析纯)购于阿拉丁试剂(上海)有限公司;氯化钡(BaCl2,分 析 纯)、硫 酸 铵 ((NH4)2SO4,分 析 纯)、PVA(Mw≈27 000)购于上海麦克林生化科技有限公司;商用纳米BaSO4购于佛山市安亿纳米材料有限公司;实验所用水均为三次水和电阻为18.2 MΩ以上的超纯水。
1.2 实验过程
1.2.1 C-N-CDs的合成
参照文献[22],将2 g柠檬酸溶解在60 mL去离子水中,加入3.2 mL乙二胺,然后200℃水热反应5 h。将反应所得溶液自然冷却至室温,使用孔径为0.22 μm的水相针式过滤器过滤后,再使用截留分子量为1 000的透析袋透析24 h,在70℃、真空下进行旋蒸,得到约5 mL的深棕色黏稠液体。
1.2.2 纳米BaSO4的制备
取不同体积(0、1、3、5、7、9 mL)的C-N-CDs,将其溶于去离子水中制备15 mL的C-N-CDs水溶液(体积分数分别为0%、6%、20%、33%、47%、60%),将0.02 mol的氯化钡溶于上述溶液,充分搅拌后得到A液。将0.02 mol的硫酸铵溶解在30 mL去离子水中,充分搅拌后记为B液。在室温下,一边充分搅拌一边将B液逐滴滴加到A液中,继续搅拌陈化120 min至反应完全,所得白色沉淀经抽滤、洗涤、干燥即得纳米BaSO4粉末样品。将由不同含量C-N-CDs(0、1、3、5、7、9 mL)的A液所制备的样品分别标记为Ba-0、Ba-1、Ba-2、Ba-3、Ba-4、Ba-5。
另取5 mL的EDTA溶于去离子水中制备15 mL的EDTA水溶液,将0.02 mol的氯化钡溶于上述溶液中,充分搅拌后得A液。除此之外的实验操作与其他样品的制备完全相同。得到的BaSO4样品标记为Ba-E。商用纳米BaSO4通过购买所得,标记为Ba-C。
1.2.3 纳米BaSO4/PVA薄膜的制备
将Ba-0、Ba-1、Ba-2、Ba-3、Ba-E、Ba-C分别与PVA颗粒按照8∶92的质量比(即0.24 g BaSO4、2.76 g PVA)均匀混合,加入10 mL去离子水后,搅拌下90℃加热处理1 h确保PVA颗粒充分溶解于去离子水中且BaSO4样品均匀分散于PVA溶液中。然后,将所得的PVA溶液全部转移到直径8 cm的培养皿中,静置1 h消除气泡,在70℃下烘干3 h即可获得BaSO4/PVA薄膜,分别标记为Ba-0/PVA、Ba-1/PVA、Ba-2/PVA、Ba-3/PVA、Ba-E/PVA、Ba-C/PVA。
1.3 测试与表征
采用捷克FEI公司的FEI/Talos L120C高分辨透射电子显微镜(HRTEM)对样品进行表征,加速电压为200 kV;采用北京普析通用仪器有限公司的MSALXD-2 X射线衍射仪对样品的结构进行检测,测试参数:辐射源为CuKα(λ=0.154 051 nm),测试电压36 kV,电流30 mA,扫描范围为5°~80°,扫描速率为 8(°)·min-1;采用傅里叶红外交换光谱仪(FT-IR,Model Bruker Vertex 70 FTIR)测定FT-IR谱图;采用马尔文公司的Zetasizer Nano ZSE激光粒度分析仪对样品表面电性进行测试;采用日本日立公司生产的F-7000型荧光光谱仪测试其荧光发射光谱及其激发光谱;采用ZEISS公司的EVO MA 15扫描式电子显微镜(SEM)对样品的微观察形貌进行表征,加速电压为30 kV;采用万能试验机(UTM-4204,Ttzh,China)对薄膜进行应力-应变测试。
2 结果与讨论
2.1 CDs的表征
2.1.1 TEM表征、荧光光谱及XRD表征
为确定C-N-CDs的形态及结构特征,对C-NCDs进行以下表征:其TEM图(图1a)显示所制备的C-N-CDs呈类球体,分散性良好;由图1a的插图可见,C-N-CDs具有明显的晶格条纹,晶格间距为0.21 nm;经过粒径统计得到粒径分布统计图,如图1b所示,C-N-CDs平均粒径为2.7 nm,粒径分布为1.3~3.9 nm,分布均匀。对C-N-CDs的荧光性能进行分析,结果见图1c。C-N-CDs的最佳激发波长为397 nm,最佳发射波长为445 nm。随着激发波长从360 nm变化为430 nm,其发射波长仍然保持在440 nm附近,仅有微弱的红移,不具备激发波长依赖效应。图1c的插图是C-N-CDs在自然光(左)与紫外光(右)下的照片,在自然光下C-N-CDs为深棕色黏稠液体,在紫外灯下C-N-CDs能发射出明亮的蓝光,这正是C-N-CDs荧光性能的表现。其XRD图(图1d)显示C-N-CDs在2θ=21°附近有一个宽的衍射峰,对应石墨碳的(100)晶面,表明C-N-CDs的碳核由结晶碳与不定型碳组成。

图1 C-N-CDs的TEM图(a)及其晶格条纹(插图)、粒径分布统计图(b)、荧光光谱及C-N-CDs在自然光与紫外光下的图像(插图)(c)和XRD图(d)Fig.1 TEM image(a)and corresponding lattice fringes(Inset),particle size distribution statistics(b),fluorescence spectra and images of C-N-CDs under natural light and UV light(Inset)(c),and XRD pattern(d)of C-N-CDs
2.1.2 FT-IR谱图及ζ电位
为分析C-N-CDs的表面特性,我们对C-N-CDs的FT-IR谱图及不同浓度水溶液的ζ电位进行表征。如图2a所示,对于 C-N-CDs,2 900~3 366 cm-1处的吸收峰对应N—H/O—H键的伸缩振动峰,来源于乙二胺中的氨基以及柠檬酸中的羟基和羧基;1 655 cm-1处的峰对应C=O键的伸缩振动峰,来源于柠檬酸中的C=O键;1 544 cm-1处的峰对应N—H的弯曲振动峰[23],来源于乙二胺中的氨基;1 384 cm-1处的强吸收峰对应C—N键的伸缩振动峰[24],表明柠檬酸与乙二胺在水热过程中发生了酰胺化反应,通过化学键相互结合[22]。对于EDTA,3 382 cm-1处的吸收峰对应O—H键的伸缩振动峰,而1 655 cm-1处的峰对应C=O键的伸缩振动峰,来源于EDTA中的羧基;1 384 cm-1处的强吸收峰对应C—N键的伸缩振动峰。由此可见,C-N-CDs与EDTA具有类似的配位原子:羧基上的氧原子与氨基上的氮原子。

图2 C-N-CDs与EDTA的FT-IR谱图(a);不同浓度C-N-CDs水溶液的ζ电位图(b)Fig.2 FT-IR spectra of C-N-CDs and EDTA(a);ζ potential diagrams of aqueous solutions of C-N-CDs with different concentrations(b)
如图2b所示,体积分数分别为6%、20%、33%、66%、100%的C-N-CDs水溶液的ζ电位分别为-5.5、-12.2、-16.3、-18.5、-23.2 mV。可见C-N-CDs在水溶液中呈电负性,且随着浓度的增加,电负性逐渐增强。C-N-CDs的电负性可能是其表面官能团羧基与羟基水解的结果。
2.2 纳米BaSO4颗粒的表征
2.2.1 外观与发光性能
纳米BaSO4颗粒的外观摄影图像如图3a所示,在C-N-CDs调控下制备的纳米BaSO4颗粒Ba-3与商用纳米BaSO4颗粒Ba-C比较,Ba-3的色泽略微变暗,这是因为BaSO4颗粒的色泽被深棕色的C-N-CDs改变,但BaSO4的高折射率极大降低了C-N-CDs的影响,Ba-3色泽变化不大。在紫外灯的照射下,如图3b所示,Ba-C并未发光,而Ba-3发射出明亮的蓝光。进一步表征Ba-3的荧光光谱,如图3c所示,发现相较于CDs,Ba-3的最佳激发峰发生蓝移且显著变宽,可能是受基质BaSO4的影响。

图3 Ba-C、Ba-3在日光灯(a)、紫外灯(b)照射下的图像;Ba-3的荧光光谱(c)Fig.3 Images of Ba-C and Ba-3 illuminated by fluorescent lamps(a)and ultraviolet lamps(b);Fluorescence spectra of Ba-3(c)
2.2.2 形貌与粒径
从图4所示的SEM图可以发现,沉淀法制备纳米BaSO4的过程中,随着反应体系中C-N-CDs浓度的增加,所制备的BaSO4样品形貌逐渐由扁平颗粒变为类球体,粒径则先减小后不变,样品Ba-3为类球体的小颗粒,而样品Ba-4、Ba-5形貌与之相似。Ba-C形貌不规则,粒径大多小于1 μm,且粒径差别较大。样品Ba-E形貌大致呈类球体,粒径较Ba-3大。

图4 (a)Ba-C、(b)Ba-0、(c)Ba-1、(d)Ba-2、(e)Ba-3、(f)Ba-4、(g)Ba-5、(h)Ba-E的SEM图像Fig.4 SEM images of(a)Ba-C,(b)Ba-0,(c)Ba-1,(d)Ba-2,(e)Ba-3,(f)Ba-4,(g)Ba-5,and(h)Ba-E
如图5所示,通过TEM图像可见各样品的形貌与SEM图像相一致。Ba-0、Ba-1、Ba-2、Ba-3由不规则颗粒逐渐变化为类球体颗粒;Ba-C为不规则的颗粒,粒径差别较大;Ba-E大致呈现类球体。通过TEM图像可统计各样品的粒径分布,如图6所示,各样品粒径大致呈正态分布,其中Ba-3有近50%的颗粒分布在30~50 nm的区间内,粒径分布较窄,而其他样品粒径分布较宽。Ba-4、Ba-5样品的粒径则与Ba-3近似。如图6i所示,将各样品的粒径分布范围及平均粒径用柱状图及折线图表示,可见Ba-0、Ba-C、Ba-1、Ba-E、Ba-2、Ba-3、Ba-4、Ba-5的粒径分布范围依次呈现先缩小后大致不变的趋势,平均粒径分别为 162.1、118.2、97.7、73.7、68.6、45.3、47.8、45.1 nm,呈先减小后不变的趋势。

图5 (a)Ba-C、(b)Ba-0、(c)Ba-1、(d)Ba-2、(e)Ba-3、(f)Ba-4、(g)Ba-5、(h)Ba-E的TEM图像Fig.5 TEM images of(a)Ba-C,(b)Ba-0,(c)Ba-1,(d)Ba-2,(e)Ba-3,(f)Ba-4,(a)Ba-5,and(g)Ba-E
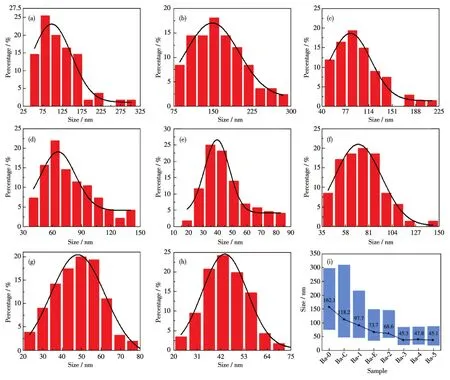
图6 (a)Ba-C、(b)Ba-0、(c)Ba-1、(d)Ba-2、(e)Ba-3、(f)Ba-4、(g)Ba-5、(h)Ba-E的粒径分布统计图;(i)各样品粒径范围与平均粒径示意图Fig.6 Particle size distribution statistics of(a)Ba-C,(b)Ba-0,(c)Ba-1,(d)Ba-2,(e)Ba-3,(f)Ba-4,(a)Ba-5,and(g)Ba-E;(i)Particle size range and average particle size diagram of each sample
2.2.3 调控机理探究
为探究C-N-CDs在反应过程中的作用,保持其他实验方法不变,在滴加反应完成后,仅进行15 min的陈化,便将样品充分洗涤、过滤、烘干,标记为Ba-3(15 min)。如图7a所示,通过标准 BaSO4、Ba-3(15 min)、Ba-3的FT-IR谱图可发现:经过120 min陈化制备的Ba-3样品与标准BaSO4的FT-IR谱图一致,在 610、983、1 081、1 117、1 186 cm-1处存在BaSO4的特征吸收峰。而Ba-3(15 min)的FT-IR谱图除了上述BaSO4的特征吸收峰外,还存在来自C-N-CDs的特征吸收峰。如图8所示,进一步对Ba-3(15 min)与Ba-3中的Ba、S、O、N元素的分布情况进行表征,可见Ba-3(15 min)中的N元素比Ba-3中的N元素含量高。TEM的元素成像可检测到样品颗粒内部,结合FT-IR谱图的测试结果,N元素含量的减少是因为反应完成后C-N-CDs无法附着在样品颗粒表面。这可能是在陈化反应初期,沉淀反应尚未完全,带负电的C-N-CDs吸附于BaSO4晶核带正电的晶面,并随着晶核的生长被镶嵌在BaSO4晶核表面,进而发挥作用;随着陈化反应的完成,BaSO4晶体没有带正电的晶面,C-N-CDs无法附着在BaSO4颗粒表面。

图7 标准BaSO4及反应15、120 min所制备的Ba-3样品的FT-IR谱图 (a);Ba-0、Ba-1、Ba-2、Ba-3、Ba-E的XRD图 (b)Fig.7 FT-IR spectra of standard BaSO4and Ba-3 samples prepared by reaction for 15 and 120 min(a);XRD patterns of Ba-0,Ba-1,Ba-2,Ba-3,and Ba-E(b)

图8 Ba-3(15 min)(a)与Ba-3(b)的TEM图像及其Ba、S、O、N元素成像图Fig.8 TEM images of Ba-3(15 min)(a)and Ba-3(b)and corresponding Ba,S,O,N element images
由图7b的XRD图可见,Ba-0、Ba-1、Ba-2、Ba-3、Ba-E均有2θ=25.84°、26.84°、28.75°、31.52°、32.72°、42.91°对应的衍射峰,属于BaSO4;另外,Ba-0、Ba-1、Ba-2、Ba-3的衍射峰逐渐减弱,表明在添加不同浓度的C-N-CDs后,BaSO4晶体的生长受到不同程度的影响,样品结晶度逐渐降低,可能是因为C-N-CDs附着在BaSO4微晶表面阻碍了晶体的生长;Ba-0、Ba-1、Ba-2、Ba-3的衍射峰逐渐变宽,表明各样品的粒径逐渐变小。
如图9a所示,Ba-0、Ba-E、Ba-1、Ba-2、Ba-3样品在陈化反应15 min时,Ba-0、Ba-E呈电中性,而Ba-1、Ba-2、Ba-3呈电负性,且电负性小幅度增强,这是由于样品颗粒表面附着有C-N-CDs;陈化反应完成时各样品大致呈电中性。如图9b所示,在滴加反应完成后,立即将Ba-0、Ba-E、Ba-1、Ba-2、Ba-3样品的反应体系静置。可见Ba-3体系中的BaSO4晶核沉积最慢,分散性最好。这是因为在Ba-3的反应体系中充满表面带有负电的C-N-CDs,使得体系中带有负电,而表面镶嵌着C-N-CDs的BaSO4晶核表面同样带有负电,在静电斥力的影响下,BaSO4微晶相互排斥,保持良好的分散状态,从而达到较小的粒径。
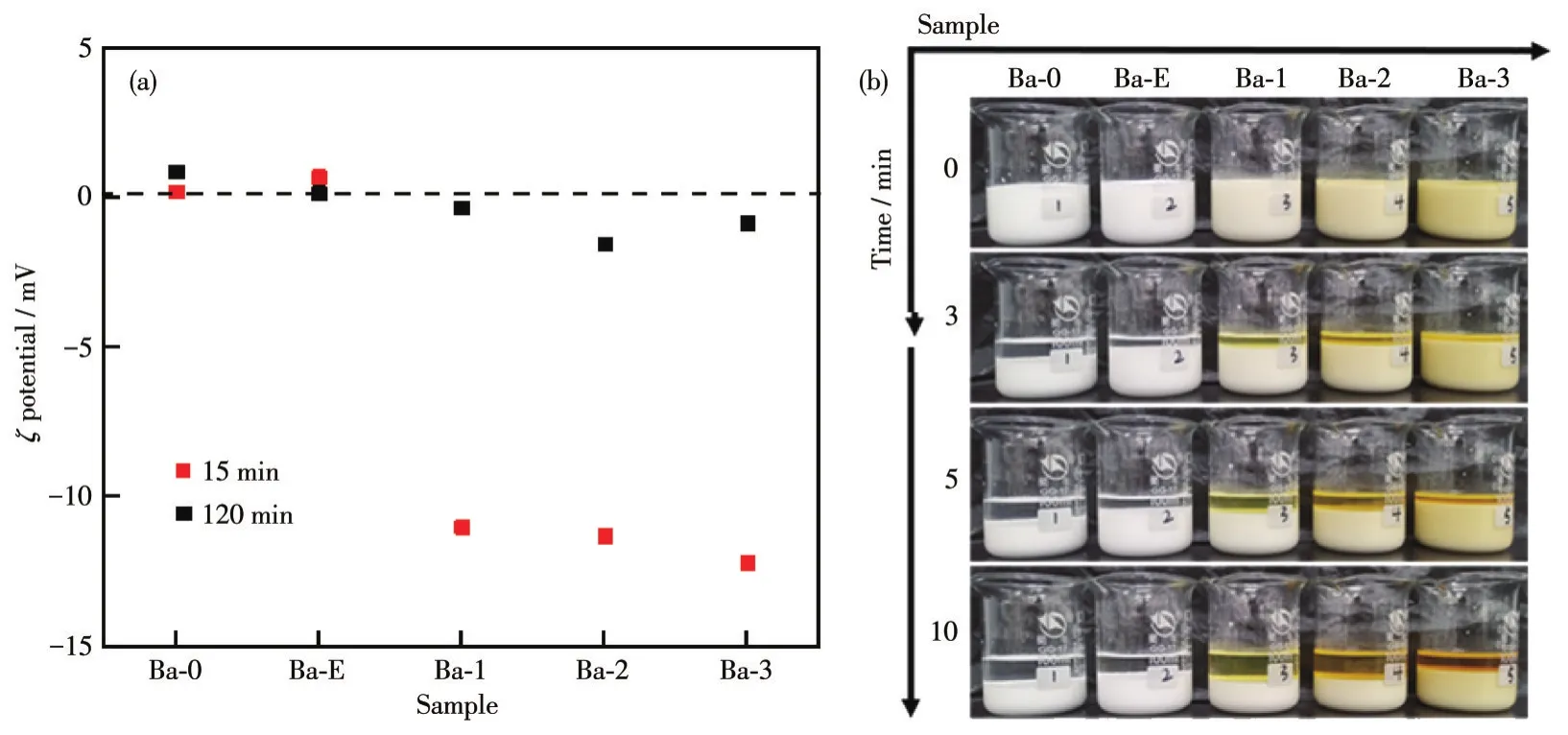
图9 Ba-0、Ba-E、Ba-1、Ba-2、Ba-3在陈化反应15和120 min时的ζ电位图(a);反应初期颗粒分散性图像(b)Fig.9 ζ potential diagram of Ba-0,Ba-E,Ba-1,Ba-2,Ba-3 at 15 and 120 min of aging reaction(a);Particle dispersion images at the beginning of the reaction(b)
2.3 高分子薄膜的性能测试
将PVA、Ba-C/PVA、Ba-E/PVA、Ba-0/PVA、Ba-1/PVA、Ba-2/PVA、Ba-3/PVA薄膜衬以黑色背景进行拍摄,如图10a所示,Ba-C/PVA、Ba-E/PVA、Ba-0/PVA、Ba-1/PVA、Ba-2/PVA薄膜中无明显的颗粒,但Ba-0/PVA薄膜中含有肉眼可见的白色BaSO4颗粒,这是BaSO4颗粒团聚所致。
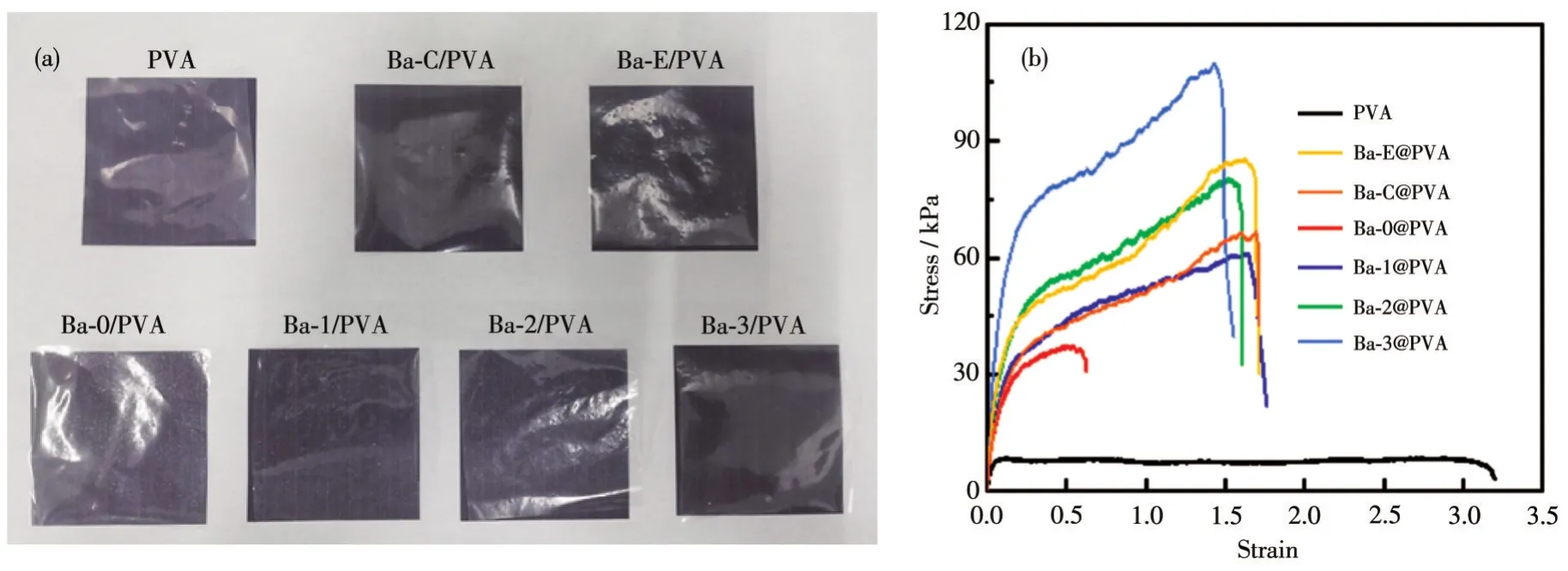
图10 PVA、Ba-C/PVA、Ba-E/PVA、Ba-0/PVA、Ba-1/PVA、Ba-2/PVA、Ba-3/PVA薄膜的实体图片 (a)和应力-应变曲线 (b)Fig.10 Real images(a)and stress-strain curves(b)of PVA,Ba-C/PVA,Ba-E/PVA,Ba-0/PVA,Ba-1/PVA,Ba-2/PVA,and Ba-3/PVA films
对PVA、Ba-C/PVA、Ba-E/PVA、Ba-0/PVA、Ba-1/PVA、Ba-2/PVA、Ba-3/PVA薄膜的力学性能进行表征,如图10b的应力-应变曲线所示。纯PVA薄膜在较低的应力下拉伸超过原本长度的3倍;Ba-0/PVA薄膜的韧性有所增强,弹性大幅度下降,但相较于添加其他样品的薄膜其力学性能最差,这是因为Ba-0粒径大、形貌不规则;Ba-1/PVA、Ba-E/PVA、Ba-2/PVA、Ba-C/PVA、Ba-3/PVA薄膜的韧性大幅度增强,弹性略微下降,力学性能逐渐增强。
2.4 机理分析
沉淀反应的推动力为溶度积(Ksp)与浓度积(Q)的差值。当Q>Ksp时有沉淀产生,这个差值越大沉淀反应速度越快;当Q≤Ksp时,沉淀反应无法进行。在本研究中,C-N-CDs与EDTA含有相同的配位原子,起到了配位剂的作用,但C-N-CDs通过表面电性与空间位阻使得制备的BaSO4粒径更小。BaSO4样品的制备过程分3个部分进行:(1)配位阶段,即A液的制备过程。此阶段通过充分搅拌使C-N-CDs与Ba2+反应形成金属配合物,Ba2+浓度被大幅度降低,而且在后续反应中只能被缓慢释放。C-N-CDs的浓度越大,越多的Ba2+被配位,但C-N-CDs浓度达到一定程度时Ba2+会被全部配位,从而导致无法通过进一步提升浓度来减小颗粒粒径。(2)滴加阶段,就是将B液逐滴滴加进A液反应的过程。此阶段B液被逐滴滴加,使得SO42-同样维持在较低浓度参与反应,这就导致反应的Q值较小,反应速度较慢,产生极小的BaSO4晶核。在这个过程中,部分C-N-CDs随着晶体的生长被镶嵌在BaSO4晶核表面,通过静电斥力的作用避免BaSO4晶核的聚集,有利于减小颗粒粒径。(3)陈化阶段,即滴加反应后的反应阶段。此阶段反应尚未完成,附着在BaSO4晶核表面的C-N-CDs通过空间位阻阻碍BaSO4晶体的生长。
3 结 论
本研究通过自下而上法,以柠檬酸、乙二胺为原料水热制备新型BaSO4粒径调控剂C-N-CDs,其空间位阻、化学性能、表面电性使沉淀法制备的纳米BaSO4颗粒粒径减小,效果优于传统的EDTA。利用C-N-CDs制备的纳米BaSO4作为无机填充物通过纳米增韧效应改善PVA薄膜的力学性能,为碳点理化性能的应用提供了新的思路。
猜你喜欢
南京航空航天大学学报(2022年4期)2022-08-30
中国食用菌(2021年10期)2021-11-04
延边大学学报(自然科学版)(2021年3期)2021-11-03
辽宁科技大学学报(2021年1期)2021-05-07
昆钢科技(2021年6期)2021-03-09
党的生活(黑龙江)(2020年10期)2020-12-18
汽车零部件(2020年9期)2020-09-27
当代陕西(2019年6期)2019-04-17
中学生数理化(高中版.高考理化)(2018年6期)2018-06-30
分析化学(2018年2期)2018-03-02
