
含二齿膦配体的二羰基铁化合物的制备及光诱导释放一氧化碳性能
2022-07-12 07:39罗佳彬郭晋忠肖志音李雪明刘小明
无机化学学报 2022年7期
罗佳彬 郭晋忠 肖志音*, 钟 伟 李雪明 刘小明
(1桂林理工大学化学与生物工程学院,桂林 541006)(2嘉兴学院生物与化学工程学院,嘉兴 314001)
0 引 言
一氧化碳(CO)是人体内一类重要的气体信号分子,能参与多种信号转导通路并发挥系列生理调节作用,具有抗炎、抗氧化、细胞保护、降低缺血再灌注损伤、减轻器官移植排异等医学研究价值[1-11]。一氧化碳释放剂(CO-releasing molecule,CORM)是一类能够以“固体形式”储存CO、并经由一定作用方式释放CO的化合物[12]。CORM为CO在体内的精准输送提供了便利,对有关CO的药用价值研究和临床应用探索具有重要意义。因此,已有大量的过渡金属羰基化合物及一些含羰基的有机物或无机化合物作为CORM被报道和研究[13-22]。
铁羰基化合物是一类经典的过渡金属羰基化合物。铁的价态、CO与铁的结合方式、非羰基配体的类型较丰富,这为功能定制的铁羰基化合物及材料的设计、合成提供了可选策略,并进一步促进了其结构多样性研究[23-24]。由于铁是人体必需的微量元素,在体内的代谢机制成熟,因此基于铁羰基化合物的一氧化碳释放剂(FeCORM)也受到了科研工作者的青睐[14,23,25-28]。我们课题组围绕FeCORM的水溶性、CO释放可控性及其生物毒性也开展了一些工作[29-35],近期的综述性论文对该方面的研究进展有较系统的总结[23]。FeCORM释放CO的速率与铁中心的价态、CO的数目及结合形式和非羰基配体的电子效应有紧密联系。一般铁中心的价态较低(如0或+1价)、CO的数目少、非羰基辅助配体的供电子能力强,则Fe—CO键强度大,使FeCORM释放CO的速率缓慢。例如,含4个CO的二价铁化合物[Fe(CO)4I2]在极性溶剂中(DMSO、H2O)几分钟内即降解完全,释放CO速率太快而难以应用[26];但以之作为前驱体制得的三羰基化合物,则稳定性显著提升,如含fac-[Fe(CO)3I3]-的离子型化合物在DMSO中的半衰期在2 h以上[31]。本研究中,通过选用3个二齿膦配体,与[Fe(CO)4I2]前驱体反应,制得了二齿膦二羰基铁化合物1~3,期望通过配体的强电子效应和差异以调节化合物的稳定性。本工作还研究、比较了这类化合物在暗处和可见光照射下的CO释放性能。
1 实验部分
1.1 试剂和仪器
通过惰性气体保护Schlenk技术以制备对水、氧气敏感的配体或配合物。反应所用的无水溶剂由Phoenix SDS5溶剂净化系统(JC Meyer)制得。1,2-双(二苯基膦酰)乙烷(dppe)和 1,3-双(二苯基膦酰)丙烷(dppp)购自上海阿拉丁生化科技股份有限公司并直接使用。N-环己基-N-(二苯基膦酰)-1,1-二苯基膦胺(Ph2PN(cyclohexyl)PPh2,PNP)[36]和[Fe(CO)4I2][32]参照文献方法合成。
实验中使用的主要仪器有红外光谱仪(Nicolet iS10,Thermo Fisher Scientific公司)、紫外可见分光光度计(Thermo EV201,Thermo Fisher Scientific公司)、核磁共振波谱仪(Varian 400-MR,Agilent公司)、X射线单晶衍射仪(Gemini X,Agilent公司)、元素分析仪(Vario EL Ⅲ,Elmentar公司)。
1.2 铁羰基化合物1~3的制备
一般制备流程如下:在氩气气氛下,向100 mL反应瓶中加入0.422 g(1 mmol)的[Fe(CO)4I2]和20 mL的二氯甲烷(DCM)。完全溶解后,将温度降至0℃(冰水浴),通过恒压滴液漏斗向反应瓶中逐滴加入含有1 mmol二齿膦配体(dppe、dppp、PNP)的DCM溶液(15 mL),滴加过程中可观察到显著的颜色转变。利用FT-IR检测反应直至前驱体反应完全(约30 min)。经减压抽滤除去沉淀物,收集母液并浓缩。粗产品用硅胶填充柱进行柱层析分离提纯(氯仿为洗脱剂)。产物经重结晶得纯品。
[Fe(cis-CO)2(dppe)I2](1)用 dppe(398 mg,1 mmol)合成,得到暗红色固体。产率:0.574 g(70%)。FT-IR(DCM,cm-1):2 035,1 990。UV-Vis(DCM,λmax/nm):348,497。1H NMR(400 MHz,CDCl3):δ8.09~7.21(m,20H,Ar-H),3.58~3.30(m,2H,CH2),3.24~2.97(m,1H,CHaHb),2.94~2.67(m,1H,CHaHb)。31P{1H}NMR(162 MHz,CDCl3):δ91.91(JP-P=29.3 Hz),49.23(JP-P=29.3 Hz)。1(C28H24FeI2O2P2)的元素分析(%)计算值(实验值):C,44.01(44.60);H,3.17(3.48)。
[Fe(cis-CO)2(dppp)I2](2)用 dppp(412 mg,1 mmol)合成,得到暗红色固体。产率:0.576 g(69%)。FT-IR(DCM,cm-1):2 036,1 983。UV-Vis(DCM,λmax/nm):361,498。1H NMR(400 MHz,CDCl3):δ8.64~6.53(m,20H,Ar-H),3.78~3.57(m,1H,CHaHb),3.53~3.31(m,1H,CHaHb),3.10~2.88(m,1H,CHaHb),2.52~2.23(m,1H,CHaHb),2.20~1.97(m,1H,CHaHb),1.31~1.12(m,1H,CHaHb)。31P{1H}NMR(162 MHz,CDCl3):δ53.21(d,JP-P=68.0 Hz),7.83(d,JP-P=68.0 Hz)。2(C29H26FeI2O2P2)的元素分析(%)计算值(实验值):C,44.76(44.20);H,3.37(3.17)。
[Fe(trans-CO)2(PNP)I2](3)用PNP(467 mg,1 mmol)合成,得到暗绿色固体。产率:0.310 g(35%)。FT-IR(DCM,cm-1):1 995。UV-Vis(DCM,λmax/nm):336,399,588。1H NMR(400 MHz,CDCl3):δ7.97~7.81(m,8H,Ar-H),7.70~7.47(m,12H,Ar-H),3.33~3.14(m,1H,NCH),1.63~1.23(m,5H,2CH2+CHaHb),1.01(dd,J=23.3,12.4 Hz,2H,CH2),0.87(dd,J=24.8,13.8 Hz,2H,CH2),0.79~0.63(m,1H,CHaHb)。31P{1H}NMR(162 MHz,CDCl3):δ114.03(s)。3(C32H31FeI2NO2P2)的元素分析(%)计算值(实验值):C,46.13(46.50);H,3.75(3.98);N,1.68(1.63)。
1.3 X射线晶体学分析
将10 mg化合物溶于1.5 mL DCM中,小心加入6 mL乙酸乙酯(EA),于-17℃下液-液扩散获得了化合物2和3的晶体。挑选质量较好的晶体,在Gemini X单晶衍射仪上通过ω-scans法采集数据。在Olex2软件中,通过ShelXT程序和SHELXL程序解析和精修结构,相关晶体学数据见表1。通过理论加氢方法获得化合物的氢原子。化合物2的晶体中存在较大的溶剂可及表面,为了获得更高质量的结构解析结果,使用Olex2软件自带的“Use solvent mask”程序屏蔽了溶剂的干扰,有关masks处理信息见其CIF文件。

表1 化合物2和3的晶体学数据Table 1 Crystallographic data for compounds 2 and 3
CCDC:2154703,2;2154704,3。
1.4 化合物1~3在DMSO中释放CO
避光释放:将化合物1(11.5 mg,0.015 mmol)溶于3 mL DMSO(5 mmol·L-1),然后将该溶液置于暗处的37℃水浴中搅拌,并每隔一定时间移取反应液,用红外光谱法监测化合物的降解释放CO情况。化合物2和3的监测过程相同。
光照释放:采用同样方法配制化合物1的DMSO 溶液(5 mmol·L-1),将其置于37 ℃水浴中搅拌,并在正上方15 cm处设一LED灯(功率2 W)照射,每隔一定时间移取反应液测试其降解释放CO情况。为考察光源波长对其降解的影响,选取了3个波段的可见光光源:红光(615~650 nm)、绿光(495~530 nm)和蓝光(450~480 nm)。化合物2和3的监测过程相同。通过线性拟合化合物的浓度(c)与光照时间(t)的关系,获得化合物在光照下的降解动力学数据。
1.5 化合物1在固相分散系、蓝光照射下降解释放CO
取化合物1(4 mg)与KBr固体(100 mg),用研钵研磨均匀(40 μg·mg-1)。在研钵正上方15 cm处设一蓝光LED灯(功率2 W)照射,每隔一定时间取样压片测试其红外光谱。
2 结果与讨论
2.1 化合物1~3的制备
前驱体[Fe(CO)4I2]具有良好的反应性,可与系列
亲核试剂(如硫醇、胺、膦、卤素等)发生取代反应得到铁羰基化合物[29-31,37-38]。本工作选用具有强供电子和配位能力的二齿膦配体(dppe、dppp、PNP)与之反应(图1),一方面减少π酸性配体CO的数目,另一方面也提高金属中心的电子密度,从而提高Fe—CO键的强度,改善目标化合物的稳定性。与P-N-P三原子共平面的PNP配体不同,dppe和dppp配体2个P原子之间的桥联烷基部分可以灵活翻转,因此这2个配体与前驱体反应时,优先形成动力学速率占优的单齿配位化合物,再形成热力学稳定性占优的二齿配位的化合物1和2;PNP配体的反应则未检测到单齿配位的中间产物。此外,dppe和dppp生成的相应产物(化合物1和2)是顺式(cis-)二羰基结构,而PNP生成的化合物3为反式(trans-)二羰基结构,这可通过晶体结构和红外表征结果确定(见下文)。这种立体构型的差异与这2类配体不同的配位特点有关。由于EA对产物的溶解度差而对反应原料的溶解性较好,因此可通过EA/DCM液-液扩散结晶获得纯品。

图1 化合物1~3的合成路线Fig.1 Synthetic routes of compounds 1-3
2.2 化合物2和3的晶体结构分析
通过X射线单晶衍射实验,确定了化合物2和3在固体晶态下的结构,其相关晶体学数据见表1,部分键长和键角参数见表2。化合物2和3的晶体结构热椭球图(Oak Ridge thermal ellipsoid plot,ORTEP)见图2和图3。每个铁中心与2个碘离子、2个羰基和1个二齿膦配体键合,形成八面体配位结构。但化合物2中2个CO为顺式构型(2个羰基碳与铁形成的夹角∠C1—Fe1—C2=89.67(14)°,接近 90°),而化合物3的两羰基为反式构型(2个羰基碳与铁形成的夹角∠C1—Fe1—C2=165.58(16)°,接近180°)。化合物2和3的Fe—C键的平均键长分别为0.178 8和0.183 1 nm,C—O键的平均键长分别为0.112 5和0.109 3 nm,与文献报道的类似化合物数据接近[26,39]。化合物2和3的∠P—M—P咬合角分别为92.33(3)°和 71.68(3)°。化合物 3 中,由 Fe1—P2—N1—P1组成的四元环片段的扭转角为1.73(12)°,接近0°,证实了该四元环片段为准平面结构,具有较强的刚性;而化合物2中2个P原子间的桥梁部分为链式结构,在溶液中的自由度较高。此外,化合物2中2个Fe—P键长相差较远(分别为0.225 71(9)和0.233 25(9)nm),且比化合物3中的2个Fe—P键要长(分别为0.221 30(10)和0.233 25(9)nm)。前者是dppp上2个P原子配位环境差异大所致,而后者则是因为P-N-P-M共平面产生了额外的电子离域效应[40]。

表2 化合物2和3的部分键长(nm)和键角(°)Table 2 Selected bond lengths(nm)and angles(°)for compounds 2 and 3
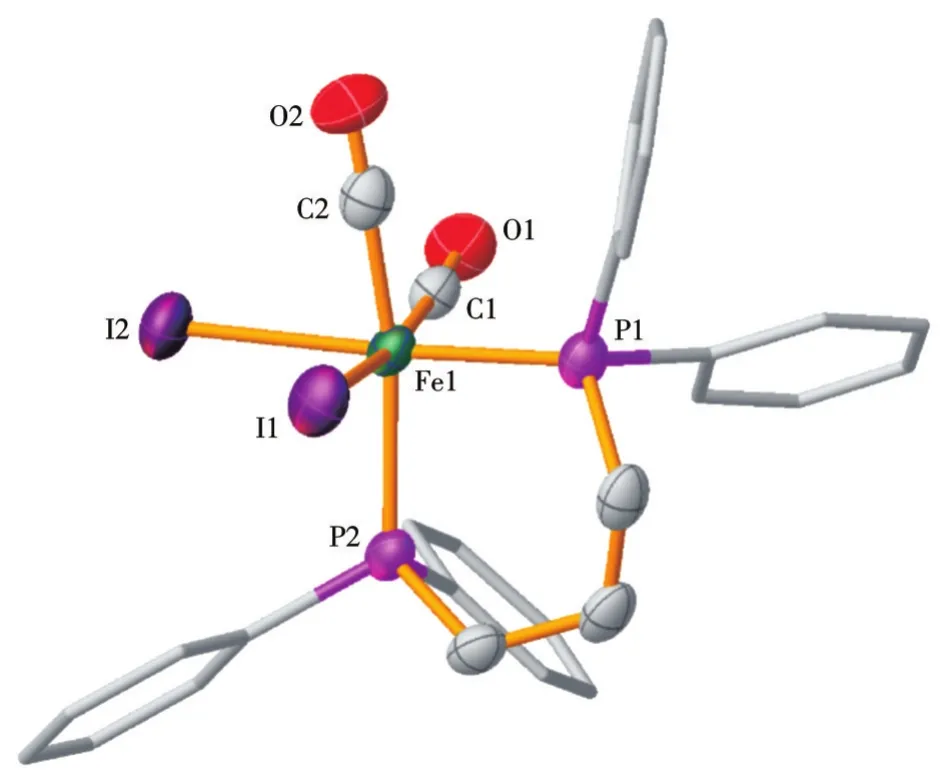
图2 化合物2的热椭球率50%的ORTEP分子结构图Fig.2 ORTEP diagram of compound 2 with thermal ellipsoids at 50% probability

图3 化合物3的热椭球率50%的ORTEP分子结构图Fig.3 ORTEP diagram of compound 3 with thermal ellipsoids at 50% probability
2.3 化合物1~3的特征红外光谱分析
铁羰基化合物在红外光谱图中具有典型的羰基振动吸收,其吸收峰位置及光谱模式能够反映铁中心的相对电子密度及CO的配位构型[38,41]。图4是化合物1~3和前驱体[Fe(CO)4I2]中羰基的特征红外光谱图。与前驱体(2 136、2 090、2 072 cm-1)相比,产物1~3的吸收峰向低波数方向显著移动:化合物1为2 035、1 990 cm-1;化合物2为2 036、1 983 cm-1;化合物3为1 995 cm-1。这归因于CO数目的减少和膦配体的强供电子能力。从羰基的特征红外光谱模式来看,化合物1和2为双峰,与顺式二羰基化合物的红外光谱相符;化合物3为单峰,与反式二羰基化合物的红外光谱相符。这与上述晶体结构分析结果一致。尽管化合物1和2都采用相同的顺式羰基立体构型,但是前者双峰的对称性要明显好于后者。这与dppp上—(CH2)3—链的构象翻转降低了其在溶液中的结构对称性有关,也与化合物的稳定性存在关联。
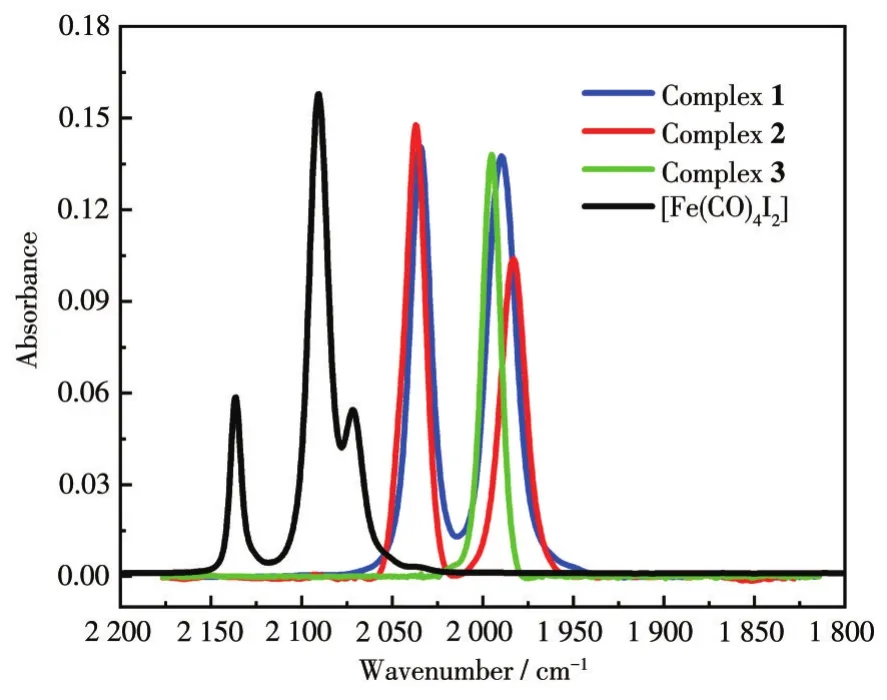
图4 化合物1~3和[Fe(CO)4I2]在DCM中的FT-IR谱图Fig.4 FT-IR spectra of compounds 1-3 and[Fe(CO)4I2]in DCM
2.4 化合物1~3的核磁共振磷谱分析
含磷化合物可通过31P{1H}NMR对其进行结构表征分析,如图5所示。与1H NMR不同,这些化合物的磷化学位移差异显著:化合物1(δ=91.91、49.23)和2(δ=53.21、7.83)均为双峰。这是由于化合物中2个P原子的配位环境完全不同(图2)。此外,化合物1和2中2个P原子间存在偶合导致双峰裂分,其偶合常数JP-P分别为29.3和68.0 Hz。化合物3则由于化学结构对称(图3),仅呈现单峰(δ=114.03)。从磷谱峰值可推断,这些膦配体供电子效应顺序为dppp>dppe>PNP。

图5 化合物1~3在CDCl3中的31P{1H}NMR谱图Fig.5 31P{1H}NMR spectra of compounds 1-3 in CDCl3
2.5 化合物1~3的吸光性能分析
通过紫外可见分光光度计检测了二齿膦配体和化合物1~3的吸光性能,见图6。膦配体在近紫外区有强吸收(峰值在253 nm附近),但与重原子铁配位后其吸收发生红移。除配体的特征吸收外,化合物1~3中还存在典型的配体与金属中心之间的电荷转移跃迁(MLCT)。化合物1(峰值348、497 nm)和2(峰值361、498 nm)在300~600 nm之间存在较强电子吸收,且化合物2比化合物1的吸收峰红移。化合物3在此区间内也有2个吸收峰(峰值336、399 nm),但要显著低于化合物1和2。这同样与膦配体的电子效应有关。此外,化合物3在588 nm附近有强吸收带,可能与Fe-P-N-P共平面带来的离域电子转移吸收有关。由图6可见,化合物1~3对可见光有较好吸收,为光诱导释放CO提供了条件。

图6 配体(a)和化合物1~3(b)在DCM中的UV-Vis谱图Fig.6 UV-Vis spectra of the ligands(a)and compounds 1-3(b)in DCM
2.6 光诱导化合物1~3降解释放CO行为
为获得热稳定性良好的铁羰基化合物,减少CO配体数目、提高非羰基配体的供电子效应是行之有效的策略。化合物1~3在暗处的DMSO溶液中的稳定性通过红外光谱法监测,结果见图7。这些化合物的羰基特征吸收在180 min内未见有显著衰减,说明其在DMSO中稳定性良好,有利于化合物的保存和运输。
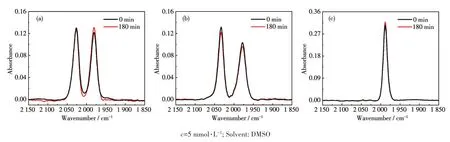
图7 化合物1(a)、2(b)、3(c)在暗处下的红外光谱图变化Fig.7 FT-IR spectral variations of compounds 1(a),2(b),and 3(c)in dark
另一方面,铁羰基化合物具有光敏性,因此可通过光诱导以促进其降解释放CO。以化合物1为例,在2 W的可见光光源(红光、绿光、蓝光)照射下,其羰基吸收峰强度随光处理时间的增加而持续衰减(图8a~8c),说明其在光诱导下分解释放CO。但不同光源下降解行为略有不同:一方面,降解速率随光源波长变短而加快,即释放CO速率为红光<绿光<蓝光。这是由于波长变短,光子能量增加而降解速率加快。另一方面,化合物1的羰基特征双峰衰减速率也不一样。红光下,双峰同等程度降低;但是在绿光和蓝光下,左峰(2 026 cm-1)的衰减要快于右峰(1 981 cm-1),且右峰向左微移。这是由于化合物1在降解释放CO的同时,产生了新物种(如蓝光照射15 min:1 989 cm-1,单峰)。经红外光谱比对(图8d),新物种不论是吸收峰位置还是光谱模式,均与化合物3一致(1 989 cm-1,单峰),由此推断其为反式二羰基物种(图9)。可见,顺式构型的化合物1,在光源能量充足的条件下(蓝光、绿光),可向反式构型转化;但在光源能量不足时(红光)则无构型转化。
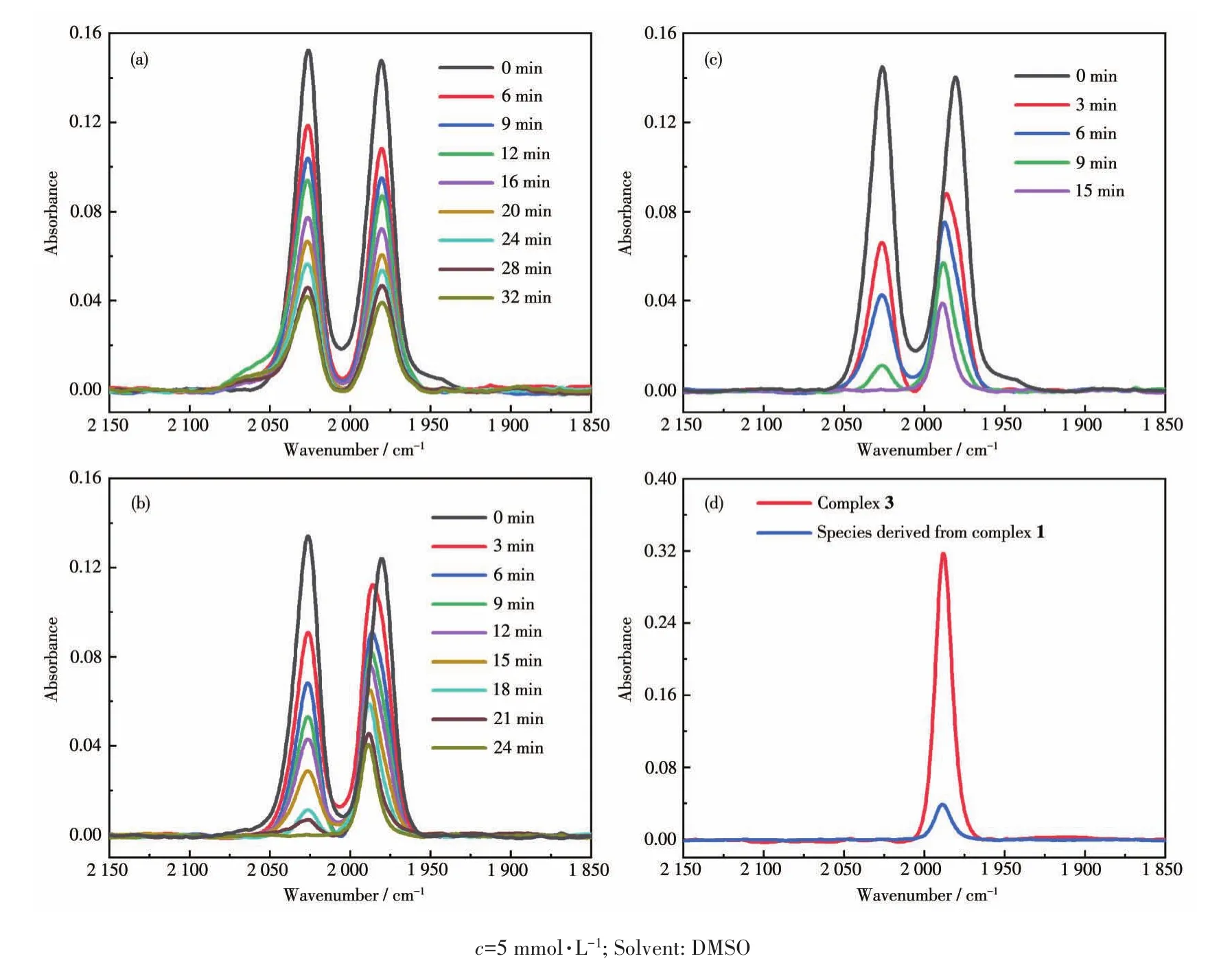
图8 化合物1在红光(a)、绿光(b)和蓝光(c)照射下的红外光谱图变化;蓝光照射15 min后的产物与化合物3的红外光谱(d)Fig.8 FT-IR spectral variations of compound 1 upon irradiation of red(a),green(b),and blue(c)lights;(d)FT-IR spectra of compound 3 and species derived from compound 1 upon irradiation of blue light for 15 min

图9 化合物1在绿光或蓝光诱导下从顺式向反式构型的转化Fig.9 Isomerization from cis-to trans-configuration of compound 1 with the induction of green or blue lights
化合物1在固相分散系(与KBr粉末研磨均匀)、蓝光照射下的降解行为也通过红外光谱进行了检测。如图10所示,与其在DMSO溶液中的光解行为相比(图8c),化合物1在固相中也可分解释放CO,但分解速率慢于其在溶液中的速率。此外,在固相中,化合物1亦可向反式构型转变(图10),不受介质的局限。

图10 (a)固态化合物1(40 μg·mg-1,KBr中)在蓝光照射下的红外光谱图变化;(b)照射40 min后形成产物的红外光谱(扣除化合物1的贡献度)与化合物3的红外光谱比较Fig.10 (a)FT-IR spectral variations of compound 1 in solid-state(40 μg·mg-1in KBr)upon irradiation of blue light;(b)FT-IR spectra of compound 3 and species derived from compound 1 upon the irradiation for 40 min(the contribution of compound 1 was subtracted from the spectrum)
化合物2在不同光源照射下的降解见图11。与化合物1类似,化合物2在绿光和蓝光下也有反式构型产物形成,但降解速率要快于化合物1。此外,化合物2在1 900 cm-1附近还有新的羰基铁物种生成。这与磷配体结构有关:由于dppp比dppe的刚性更弱,化合物2自由度更大,在光照下降解更快且产物更复杂。

图11 化合物2在红光(a)、绿光(b)和蓝光(c)照射下的FT-IR谱图变化Fig.11 FT-IR spectral variations of compound 2 upon irradiation of red(a),green(b),and blue(c)lights
图12a~12c是化合物3在这些光源诱导下的降解释放CO行为。相较于化合物1和2(图8和11),化合物3的降解速率更缓慢,且不产生其他铁羰基中间体。图12d是化合物3的浓度与光照时间之间的线性拟合关系(R2=0.99),表明该化合物的光降解过程符合零级反应动力学,其表观动力学常数(kobs)大小分别为 0.066、0.079 和 0.139 mmol·L-1·min-1。显然,其降解速率随光能量增大而加快。
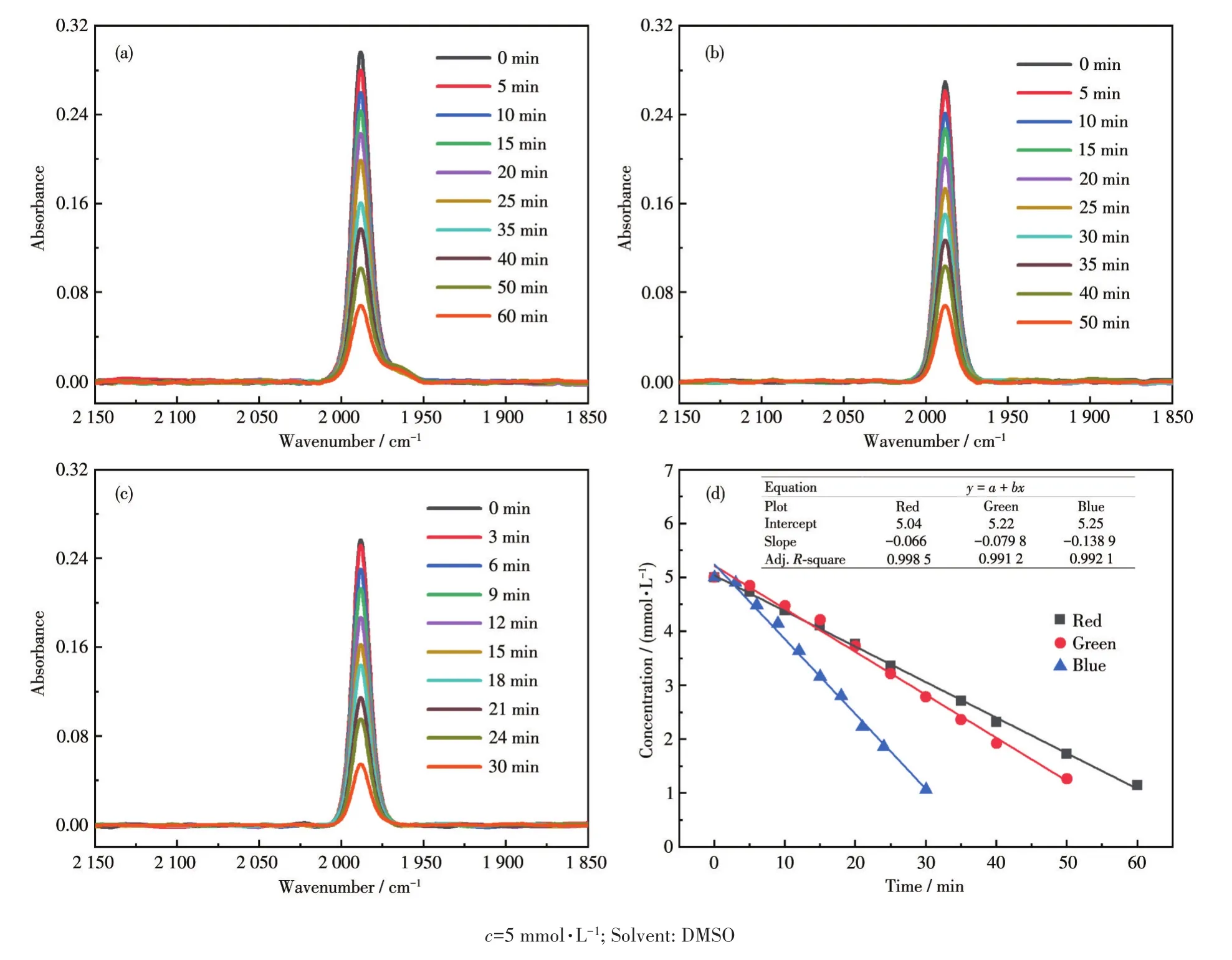
图12 化合物3在红光(a)、绿光(b)和蓝光(c)照射下的FT-IR谱图变化;(d)化合物3的浓度与光照时间之间的线性拟合关系Fig.12 FT-IR spectral variations of compound 3 upon irradiation of red(a),green(b),and blue(c)lights;(d)Linear fitting relationship between the concentration of compound 3 and the irradiation time
3 结 论
(1)通过二齿膦配体(dppe、dppp、PNP)与前驱体[Fe(CO)4I2]之间的配位取代反应制得了3个二羰基铁化合物1~3,并通过FT-IR、UV-Vis、NMR、X射线单晶衍射、元素分析等对其结构进行了表征,确定了化合物2、3的立体构型。
(2)红外光谱监测表明,化合物1~3在暗处下有良好稳定性,而在可见光(红光、绿光、蓝光)照射下均可降解释放CO,其光稳定性与可见光能量、化合物的结构有关:波长越短、光能量越大,降解越快;膦配体的刚性较强,或与铁中心存在离域电子效应,则化合物的光稳定性更好。
(3)在绿光和蓝光照射下,化合物1和2可以克服能垒,从顺式构型转变为反式构型,而红光下则因能量不足难以构型转化。在上述光照条件下,反式构型的化合物3未见其向顺式构型转化。
猜你喜欢
天津农学院学报(2022年2期)2022-08-05
渤海大学学报(自然科学版)(2022年1期)2022-07-25
云南大学学报(自然科学版)(2022年3期)2022-05-25
食品界(2022年4期)2022-04-25
现代家长(2019年2期)2019-03-18
科技创新导报(2016年30期)2017-03-15
意林·少年版(2016年11期)2016-09-10
祝你幸福·知心(2016年3期)2016-03-29
健康博览(2015年12期)2015-12-02
学苑创造·A版(2009年5期)2009-06-29
