
CRISPR/Cas9 基因编辑原理、发展及应用
2022-07-01 02:52:14董乐杨笑星佟广香闫婷孙志鹏徐欢刘天奇匡友谊
水产学杂志 2022年3期
董乐,杨笑星,佟广香,闫婷,孙志鹏,徐欢,刘天奇,匡友谊
(1.中国水产科学研究院黑龙江水产研究所,黑龙江 哈尔滨150070 2.上海海洋大学水产与生命学院,上海 201306)
自20 世纪70 年代重组DNA 技术发展以来,有关位点特异性核酸酶(Site-specific nuclease,SSN)的基础理论研究取得了突破性进展,使精准基因编辑技术快速普遍应用于各个领域。SSN 为目的基因的精准编辑提供了有效的特异性编辑方法[1,2]。较早被广泛应用于基因工程技术中的两种基因编辑技术是锌指核酸酶(Zine finger endonuclease,ZFN)和转录激活因子样效应物核酸酶(Transcription activator-like effector nuclease,TALENs)。这两种编辑方法原理相同,皆是DNA 结合蛋白与核酸内切酶FokⅠ两个模块的融合,之后对特定靶序列双链进行切割。然而这两种核酸酶存在结构复杂、不易操作、耗时长、易脱靶[3-6]等缺点,阻碍了其发展。随着2012年Jinek[7]等根据细菌的一种获得性免疫系统改造而成的成簇规律间隔的短回文重复序列(Clustered regulatory interspaced short palindromic repeat,CRISPR)介导的位点特异性人工核酸内切酶Cas9系统,在原核、真核生物的基因组编辑中取得成功而正式走进科研人员的视野[8]。CRISPR/Cas9 作为一种全新的技术具有在基因组中定点编辑的能力,具有精准、高效、制备简单等优势而被广泛发展和应用[9]。本文综述了CRISPR/Cas9 的历史来源、结构组成、作用机理、技术发展和应用等最新研究成果,旨在为初学者提供CRISPR/Cas9 基因组编辑系统的基础知识。
1 CRISPR/Cas9 的来源
CRISPR/Cas9 自发现到在哺乳动物中应用经历了近30 年的漫长研究过程(图1)。1987 年Ishino等在研究大肠杆菌(Escherichia coli)的碱性磷酸酶基因时,首先发现CRISPR 下游有被间隔序列间隔的重复序列。后来大量的细菌基因组测序,发现这种序列广泛存在于细菌与古细菌中[10]。2002 年Mojica 等[11]和Jansen 等[12]将间隔排列的串联重复序列命名为成簇规律间隔的短回文重复序列(CRISPR),同时也发现了Ⅰ型、Ⅱ型和Ⅲ型3 种类型的CRISPR 簇相关的Cas 蛋白,但其生物学功能未知。2005 年经过深入研究发现,间隔序列来源于外源入侵的DNA,推测CRISPR/Cas 可能与细菌自我保护不被外源遗传物质影响的免疫系统有关[13-15];2007 年,Barrangou R 等[16]在嗜热链球菌(Streptococcus thermophilus)中证实了CRISPR/Cas 具有获得抵抗外源噬菌体侵入的能力;2008 年Marraffini 等[17]在葡萄球菌(Staphylococcus spp.)中证明了这种获得性抵抗外源菌体的能力。Deveau 等[18]发现了原间隔序列邻近基序(protospacer adjacent motif,PAM)为CRISPR/Cas 发挥作用提供了靶标。这一防御系统的发现引起了科学家对其在基因组中定点编辑的浓厚兴趣。2012 年,Sternberg 等[19]发现II 型CRISPR中仅需一个Cas9 蛋白即可完成靶序列的编辑,在体外使用RNA 实现了DNA 目的序列的识别和剪切;2013 年Cong 等[20]和Mail 等[21]首次在真核生物中成功应用CRISPR/Cas9 进行了基因编辑。目前,CRISPR/Cas9 已逐步应用到了包括原核生物、真菌以及动植物的基因组精准编辑,成了基因组编辑中的“明星”技术。

图1 CRISPR/Cas 系统研究进展示意图Fig.1 Schematic diagram of the research progress on the CRISPR/Cas system
2 CRISPR/Cas 结构
从最初发现CRISPR/Cas 间隔排列的串联重复序列[10],并证明其广泛存在于细菌(40%)与古细菌(90%)中[22],到将带有回文结构的重复序列命名为CRISPR 序列[11,12],无一不在逐步揭开其神秘面纱。CRISPR/Cas 由CRISPR 簇和Cas 蛋白组成,CRISPR簇主要由三部分构成(图2):一个前导序列(Leader)、数个高度保守的重复序列(Repeat)和多段间隔序列(Spacer)组成[23,24]。其中Leader 序列位于CRISPR簇的上游,是CRISPR 簇的启动子序列[23];Repeat 序列一般为21~48 bp,含有回文序列,故能形成发卡结构;Spacer 区是入侵的外源DNA 序列[25]。
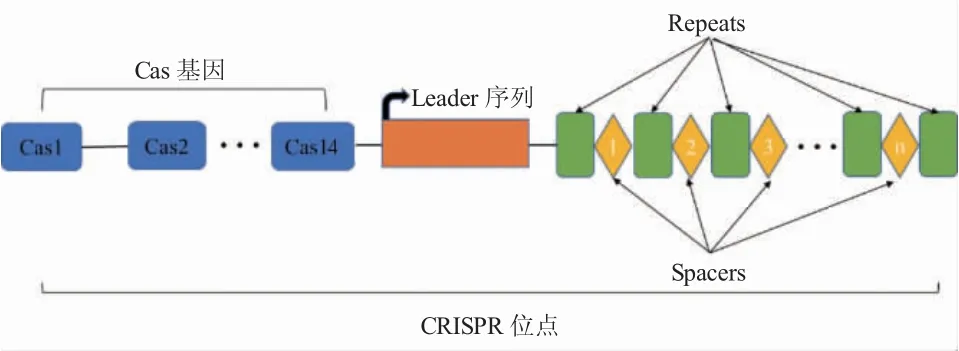
图2 CRISPR 位点结构示意图Fig.2 Schematic diagram of CRISPR structure
CRISPR 簇侧翼附近是系列CRISPR 相关蛋白基因座(CRISPR-associated),即Cas 蛋白基因[22,25],其编码产物均含有与核酸作用的功能结构,目前已发现有Cas1-Cas14 等多种类型[26-28]。Cas 蛋白基因与CRISPR 共同作用组成CRISPR/Cas 系统,完成基因的精准高效编辑。根据Cas 蛋白的种类和序列,CRISPR/Cas 可分二类六型[26-31](Ⅰ-Ⅵ),Ⅰ型、Ⅲ型和Ⅳ型需要几个Cas 效应蛋白组成的复合体,Ⅱ型、Ⅴ型和Ⅵ型仅需单个Cas 效应蛋白;其中Ⅱ型CRISPR/Cas 只需一个Cas9 蛋白,便可有效切割双链DNA。正是这种简单高效的优点给予了Ⅱ型CRISPR/Cas9 强有力的发展空间。
3 Ⅱ型CRISPR/Cas9 在细菌中的作用原理
Ⅱ型CRISPR/Cas9 与Ⅰ型和Ⅲ型两种系统在转录、加工以及与核酸酶在剪切特定目的序列的协同作用上截然不同[16],目前Ⅱ型系统的研究最为广泛和深入。CRISPR/Cas9 介导的基因编辑主要分为三个阶段(图3):首先获取外源DNA,其次CRISPR位点的转录与加工,最后是CRISPR/Cas9 切割外源DNA[32]。

图3 II 型CRISPR-Cas9 抵御外源噬菌体的获得性免疫机制,引自吴言等[39]Fig.3 The process of adaptive immune mechanism of CRISPR-Cas9's II against foreign phages,from Wu Yan et al[39]
第一阶段为获取外源DNA 序列,将外源基因的原间隔序列(protospacers)整合到宿主染色体的CRISPR 序列前导区的近端(如第一个spacer),实现标记功能。当质粒或噬菌体首次入侵携有CRISPR的个体时,在Cas1 和Cas2 的帮助下宿主首先识别入侵核酸的PAM序列,切割临近PAM 序列的外源DNA 作为原间隔序列。临近间隔序列3’端的3 nt较保守,称为PAM 序列,通常为5’-NGG-3’[33,34],同时PAM区也是该系统的剪切位点。第二阶段,当外源DNA 再次入侵时,在Leader 的调控下,CRISPR 位点被转录成RNA 前体(Pre-CRISPR RNA,pre-crRNA),同时重复序列(repeats)被转录成反式激活RNA(Trans-activating crRNA,tracrRNA),并由Cas9 和双链RNA 特异性RNase III 核酸酶对pre-crRNA 进行加工[35],生成仅含有一个间隔序列(入侵的外源序列)的成熟crRNA,该阶段Cas9 基因也被转录和表达。在crRNA、tracrRNA 和Cas9 核酸酶形成复合体crRNP 的作用下对入侵的外源DNA 进行剪切编辑[21,36],起到自我免疫防护。第三阶段是对外源DNA 的免疫编辑,复合体crRNP 识别并通过碱基互补配对结合到与间隔序列(crRNA)互补的DNA 链上,使互补链与crRNA 杂交,然后由Cas9中的HNH 核酸酶结构域剪切crRNA 的互补链,Cas9 的另一个RuvC 活性位点剪切非互补链,从而产生双链DNA 断裂(Double strand breaks,DSBs)[7],经细胞的非同源末端连接(non-homologous end joining,NHEJ)或同源介导的双链修复(homology directed repair,HDR)对DNA 进行修复[32,37],实现对外源DNA 的切割。
综上,当使用CRISPR/Cas9 对组织、细胞或者胚胎进行基因组编辑时(图4),可通过体外合成单引导RNA(Single guide RNA,sgRNA),混合Cas9 mRNA 或蛋白直接转染到细胞系或组织中[38],对目标序列进行双链切割,经NHEJ 和HDR 机制修复,即可实现基因组靶向编辑。

图4 CRISPR/Cas9 系统介导的基因组编辑,引自沈阳坤等[ 40]和Alexander Hruscha 等[41]Fig.4 Genome editing mediated by CRISPR/Cas9 system,from Shen Yangkun,et al[ 40] and Alexander Hruscha et al[41]
4 CRISPR/Cas9 的技术发展
2012 年Jinek 等[7]将crRNA 和tracrRNA 两者连接在一起,形成新的单引导RNA(Single guide RNA,sgRNA),且打靶效率与双引导RNA 并无显著改变。与野生型CRISPR/Cas9 相比,这个新型的CRISPR/Cas9 更加容易构建,只需一个引导RNA 和Cas9 核酸酶mRNA 同时注入受精卵,便可快速获得定向突变个体。该系统不仅可以靶向编辑单个基因,还可以多个sgRNA 共同使用完成多个位点的靶向突变,这在Yin 等[42]的研究中得到了很好的验证。Yin 等[42]应用多个sgRNA 在斑马鱼(Danio rerio)上获得了酪氨酸酶和胰岛素受体a 和b 的多重双等位基因失活,得到了色素沉着和葡萄糖稳态的缺陷表型。自2013 年成功将CRISPR/Cas9 基因编辑技术应用到真核生物和细胞以来[20,21],该技术取得了显著进展。
4.1 提高CRISPR/Cas9 特异性
相比于TALEN,CRISPR/Cas9 提高了靶向编辑特异性,但仍存在一定比率的脱靶效应,这将妨碍其在疾病治疗、基因功能研究上的应用。主要从改进sgRNA 和Cas9 核酸酶两方面着手以降低其脱靶率、提高特异性。在sgRNA 的改进上,Kim 等[43]和Fu 等[44]的研究分别显示延长或截短2~3 个nt 的sgRNA 能降低碱基错配率,提高靶向修饰的特异性。John 等[45,46]分析了1 841 个sgRNA 的基因编辑结果,构建了sgRNA 活性的预测模型[45]。根据该模型创建了人类和小鼠全基因组CRISPR/Cas9 sgRNA文库,在分析了数千个sgRNA 非靶标活性的基础上,开发了非靶标位点预测模型[46],改进了人类和小鼠sgRNA 的设计,以提高其活性和靶点修饰的专一性。
除了sgRNA,Cas9 核酸酶的结构也显著影响基因编辑的准确性。Cas9 蛋白通过HNH 和RuvC 两个功能结构域实现双链DNA 的切割,增加脱靶风险,因此学者对Cas9 进行了修饰。突变Cas9 两个核酸酶结构域中的一个会产生缺刻Cas9(Nickase Cas9,nCas9),该核酸酶只切割一条DNA 链[7,33]。两个nCas9 可以靶向相邻的DNA 位点而引起DSBs[47];因该过程中靶位点长度增加,产生的单个缺口将由高保真碱基切除修复机制修复,从而在不牺牲靶位点切割效率的前提下,将非靶标活性降低了50~1 000 倍[47,48]。与单结构域的失活相比,2013 年Qi 等[49]突变了Cas9 核酸酶中的两个功能结构域,让其缺乏核酸酶活性而保留其DNA 结合活性,形成了Dead Cas9(dCas9)。在不切割目标DNA 的情况下dCas9 介导特定位点的遗传和表观遗传调控,dCas9与sgRNA 共同作用时将会抑制靶基因的转录[49,50],进而大幅降低脱靶率。2014 年,在dCas9 的基础上开发的新型基因编辑工具提高了对目的位点编辑修饰的特异性。Guilinger 等[51]将dCas9 与Fok I 限制酶融合形成二聚体fCas9,经一对sgRNA 引导进行基因组修饰,其特异性要比野生型Cas9 高140倍以上,与双nCas9[47]效率相似。
4.2 CRISPR/Cas9 PAM 序列修饰
与入侵DNA 序列中同crRNA 靶序列连接临近2~5 nt 的PAM 序列决定着CRISPR/Cas9 靶点的识别,也可影响CRISPR/Cas9 介导的基因组编辑的特异性[52]。现已知存在多种PAM序列,如化脓性链球菌(Pyogeniccoccus spp.)的NGG PAM[7],嗜热链球菌(Streptococcus thermophilus)的NGGNG 和NNAGAAW PAM[32,33]及脑膜炎奈瑟菌(Neisseria meningitidis)的NNNNGATT PAM[53]等。CRISPR/Cas9 介导的DNA 切割依赖于PAM 的方式限制了基因组中靶位点的频率,NGG PAM 和NAG PAM[54]在基因组中每8 个核苷酸可以找到一个靶位点,NGGNG PAM和NNAGAAW PAM,每32 个和256 个核苷酸才能找到一个靶位点。
为扩充靶位点的数量,Nishimasu 等[55]通过基因工程方法创造了spCas9-NG 基因编辑器,将spCas9的PAM区序列从NGG 扩充至NG;Hu 等[56]采用人工进化方法,创建出xCas9,将spCas9 的PAM 区从NGG 扩充至NGN、GAA 和GAT。Kim 等[57]通过大规模的靶位点评估了野生型spCas9、xCas9 和sp-Cas9-NG PAM区靶位点的兼容性、脱靶率和编辑效率,发现xCas9 对靶位点的错配容忍性最低,而sp-Cas9-NG 具有最广泛的PAM区兼容性,相对于sp-Cas9,xCas9 和spCas9-NG 可以编辑前者不能编辑的基因。他们在此基础上进行优化并建立了xCas9和spCas9-NG 靶位点序列活性预测模型。Walton等[58]开发了SpRY 基因编辑器,将spCas9 的PAM区进一步扩充至NRN 和NYN,做到了几乎不依赖于PAM区序列进行基因编辑。
4.3 CRISPR/Cas9 介导的基因敲入
CRISPR/Cas9 不仅介导基因敲除,同样可以介导基因定向插入,CRISPR/Cas9 在产生DSB 后,可通过同源重组修复来精准且理想的达到基因修饰等目的,因此可根据需要设计sgRNA 和同源供体,以实现供体的精准敲入(图4)。例如镰刀型细胞贫血症(Sickle-cell disease,SCD)是由于单碱基突变造成。Dewitt 等[59]结合了Cas9 与sgRNA 组成的RNP复合物和单链DNA 寡核苷酸供体(ssODN),替换了SCD 患者造血干细胞HBB 突变基因中的单碱基,并将体外培养的单碱基替换的细胞植入小鼠体内存活了16 周。CRISPR/Cas9 介导的基因敲入不仅可以定点插入单碱基和小片段,还可进行大片段的精准敲入。Bai 等[60]报道了一种斑马鱼长单链DNA 模板(zLOST)。将300 nt 的zLOST 经HDR 途径敲入酪氨酸酶(Tyr)位点,结果显示98%的白化胚中至少有一个色素沉着,超一半出现少量色素沉着。该方法精准有效地修复了白化病症。Shy 等[61]在小鼠胚胎干细胞Lef1 位点介导插入了1 974 bp 的片段,尽管只有0.4%的插入效率,但也为大片段的敲入提供了案例。CRISPR/Cas9 系统介导的基因敲入同样存在脱靶问题,为了提高其精度和外源DNA 的整合效率,有研究者将sgRNA 靶位点序列加在800 bp 的同源臂两端(图5),建立了同源臂介导的末端连接(HMEJ)技术,并在小鼠胚胎及成体肝细胞中进行同源重组修复,结果显示HMEJ 介导的同源重组修复效率显著提高[62]。

图5 HMEJ 介导的基因敲入示意图,引自Yao[62]等Fig.5 Schematic diagram of HMEJ -mediated gene knock-in,from Yao et al[62]
虽然CRISPR/Cas9 可采用同源重组的方式实现基因的定点敲入,但较低的同源重组效率和DNA双链断裂引起染色体易位、重排等风险阻碍了该技术的应用。2019 年Strecker J 等[63]在蓝细菌中发现了一种Tn7 转座酶,它的三个亚基(tnsB,tnsC,tniQ)可以与CRISPR 效应物Cas12k 关联,形成CRISPR-associated transposase(CAST),开发了shCAST 系统,在大肠杆菌中实现了2.5kb 的DNA片段插入,且成功率高达80%,远超基于同源重组的CRISPR 方法。同时Klompe S E 等[64]也报道了CRISPR/Cas 与类Tn7 转座子联合介导的基因插入系统(Cascade),评估了将其应用于基因组编辑的潜力。两个系统相比,shCAST 系统更小并具有方向性,而Cascade 系统更加特异但插入是随机的,在疾病和癌症的治疗中产生了不确定性。基于转座的定向插入系统进行基因修复与现有的利用同源重组的编辑方式相比,在安全性和效率上都更具优势。
4.4 CRISPR/Cas9 介导的单碱基编辑
基于CRISPR/Cas9 可实现基因的敲除和转入及基因的单碱基编辑。2016 年David Liu 实验室用具有单一切割活性的nCas9 蛋白与胞嘧啶脱氨酶构建了单碱基编辑器CBE(Cytosine base editors,CBE);2017 年构建了ABE(Adenine base editor,ABE)单碱基编辑器,CBE 和ABE 可分别利用胞嘧啶脱氨酶或经过改造的腺嘌呤脱氨酶对靶位点上一定范围的胞嘧啶(C)或腺嘌呤(A)进行脱氨基反应,经DNA 修复和复制,从而在不产生DNA 双链断裂的情况下完成C>T(G>A)和A>G(T>C)碱基的自由转换[65,66]。哺乳动物细胞中存在尿嘧啶DNA 糖基化酶(Uracil DNA glycosylase,UDG),导致细胞内的碱基错配修复途径(Base excision repair,BER)逆转U·G 中间产物返回到C·G 碱基对(图6)。为此,该实验室将尿嘧啶DNA 糖基化酶抑制剂(Uracil DNA glycosylase inhibitor,UGI)加在了CBE 系统上,以此来提高CBE 编辑器在哺乳动物中的编辑效率。在实际应用中,Chadwick 等[67]利用CBE 编辑器在成年小鼠肝脏的PCSK9 基因中提前产生了终止密码子,编辑后小鼠的PCSK9 和胆固醇水平显著降低。随后Ryu 等[68]将ABE mRNA 和sgRNA 显微注射到小鼠胚胎中,在Try 基因中引入了白化病点突变,获得了具有白化病表型的Try 突变小鼠。这两种单碱基编辑系统的问世不仅是精准基因编辑领域的重大突破,同时对治疗许多点突变的遗传病有重要的意义,为众多遗传疾病的治疗提供了新的工具和思路。
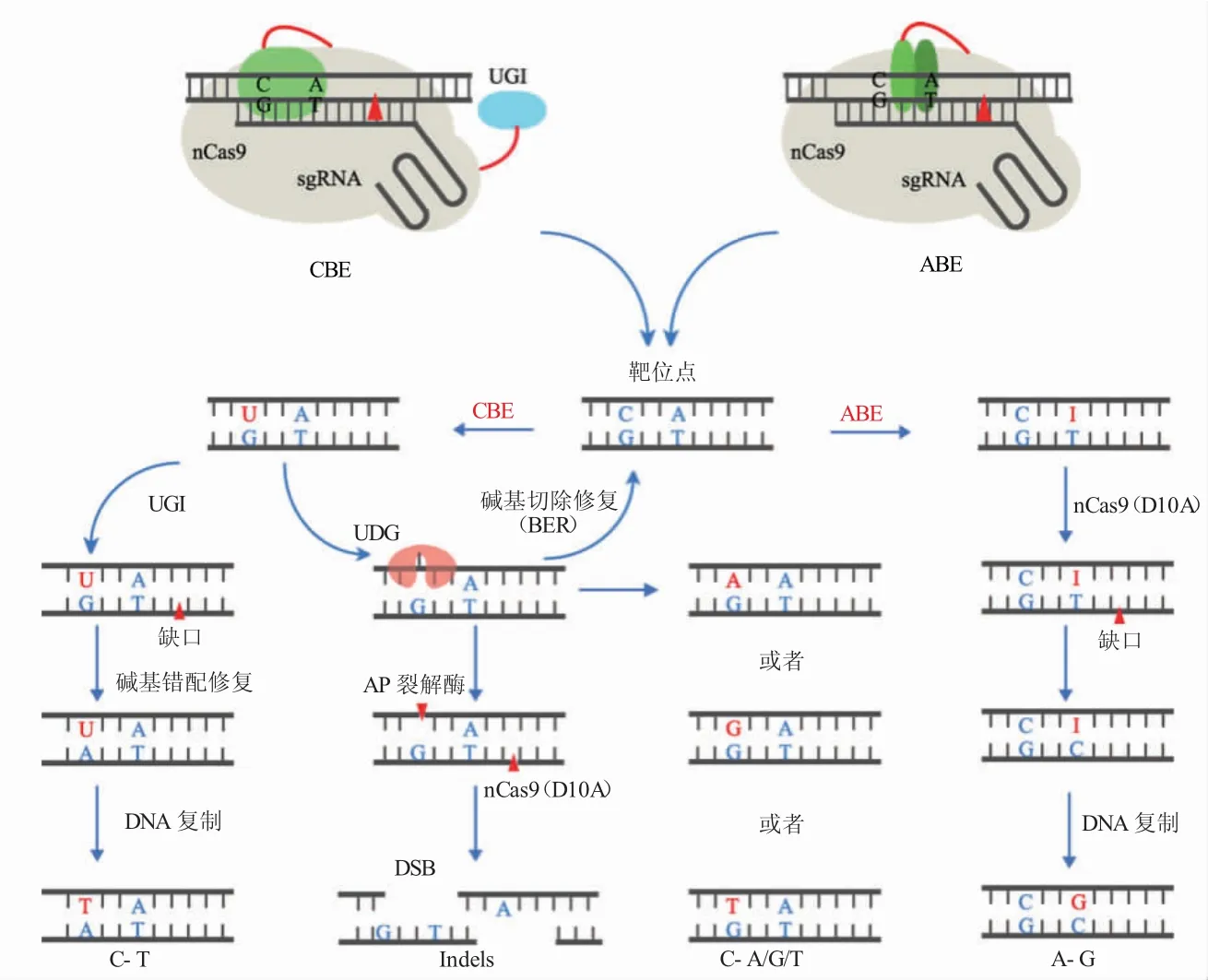
图6 CBE 和ABE 工作示意图,引自宗媛和高彩霞[69]Fig.6 Working diagram of CBE and ABE,from Zong Yuan and Gao Caixia[69]
ABE 和CBE 单碱基编辑器虽然能进行A>G(T>C)和C>T(G>A)的替换,但不能同时进行这2种类型碱基的修饰。为解决这一问题,Zhang 等[70]将人类胞嘧啶脱氨酶hAID-腺嘌呤脱氨酶-Cas9n(SpCas9 D10A 突变体)融合在一起,形成了新型双碱基编辑器A&C-BEmax。它可以在靶序列上实现C>T 和A>G 的高效转换,同时RNA 脱靶水平大幅降低,提高了C>T 的编辑效率,而A>G 的效率略有降低。随后Sakata 等[71]将来源于七鳃鳗(Petromyzon marinus)的胞嘧啶脱氨酶PmCDA1 和鼠源的胞嘧啶脱氨酶rAPOBEC1 分别与腺苷脱氨酶(TadA)融合到nCas9 的C 端和N 端,开发了双碱基碱基编辑器,成功在哺乳动物细胞中实现了A 和C 的同时突变,A 和C 的编辑效率分别高达40%和50%。最后Grünewald 等[72]采用了更短的腺苷脱氨酶(TadA)融合在N 端,将来源于七鳃鳗的胞嘧啶脱氨酶Pm-CDA1 融合到nCas9 C 端,开发了能同时编辑A 和C 的SPACE 系统。该碱基编辑器C>T 平均编辑效率和单个CBE 效率相当,A>G 平均编辑效率与单个ABE 效率相比略有下降,但是SPACE 双碱基编辑效率明显高于ABE+CBE 的共同作用,与Sakata等[71]的研究结果一致。
虽然采用CRISPR/Cas 可方便、高效地进行基因敲除、敲入和单碱基编辑,但仍不能采用单一的工具同时实现上述功能。2019 年哈佛大学David Liu实验室通过将M-MLV 逆转录酶融合到nCas9,开发了Prime Edit 基因编辑工具[73],可实现12 种单碱基替换,最多44 bp 的插入和80 bp 的删除;该系统通过在sgRNA 3’端加上一段用于逆转录的模板DNA,形成可编程的pegRNA。由pegRNA 引导融合蛋白结合到靶位点,nCas9 切割pegRNA 的非互补链,通过逆转录酶将pegRNA 3’模板DNA 逆转录到基因组中,实现无需供体DNA 的碱基替换、插入和删除。与其他基因编辑工具相比,对目的基因进行碱基替换、小片段插入和删除更有优势,该工具能使基因编辑更加高效精准。
5 CRISPR/Cas9 基因编辑的应用
CRISPR/Cas9 组成简单、易操作和编辑精准高效,使其在功能基因研究、种质培育、生物模型构建和基因治疗等研究领域有无可比拟的优势。
5.1 CRISPR/Cas9 在功能基因研究中的应用
随着模式生物基因组测序的逐渐完善健全,利用CRISPR/Cas9 在基因组中同时进行多靶位点的编辑成为可能,能通过全基因组功能筛选鉴定在目标表型中起重要作用的基因。有研究显示:将功能缺失突变引入不同基因的编码外显子中,能够在人细胞中执行强大的阴性和阳性筛选能力[74,75]。相比于早期使用RNAi 的修饰效率低下、脱靶严重以及仅限于转录而言,Cas9 和sgRNA 联合介导的筛选可提供更高的筛选敏感性和专一性,并可设计为靶向几乎任何DNA 序列[74]。Cas9-sgRNA 方法在靶基因位点产生Indels,能导致基因功能的完全丧失,而RNAi 方法只导致基因表达抑制。因此,当靶向基因相同时,CRISPR/Cas9 系统可产生比RNAi 更明显的表型,这更容易鉴定相关基因[76]。
现已证明CRISPR 介导的转录抑制(CRISPRinterference,CRISPRi)和激活(CRISPR-activation,CRISPRa)是筛选功能基因组的强大工具[77,78]。通过突变使Cas9 的HNH 和RuvC 结构域失活,形成只拥有DNA 结合活性,而无核酸酶活性的缺陷Cas9(dCas9),dCas9 和sgRNA 组成的CRISPRi 并非通过在DNA 双链断裂后引入插入缺失来使基因失活,而是特异性地靶向启动子区域抑制基因的转录[49,79,80];而CRISPRa 采用dCas9 与不同转录激活域进行融合,通过化脓性链球菌sgRNA 或募集额外转录激活域以上调靶基因表达的特殊sgRNA 定向至启动子区域[77,81,82],以此激活基因的表达,进而研究基因的功能。
5.2 CRISPR/Cas9 在生物育种中的应用
基因编辑已广泛运用于水产动物、畜禽和植物的种质创制上。Wargelius 等[83]通过CRISPR/Cas9技术成功突变了大西洋鲑(Salmo salar)中脊椎动物生殖细胞存活的必需因子Dnd,获得了不育的大西洋鲑,解决了鲑鱼养殖中逃逸对野生种群遗传干扰的问题。Ma 等[84]通过基因编辑突变了感染草鱼(Ctenopharyngodon idella)出血病毒GCRV 的必需基因gcJAM-A,从而使草鱼获得了抗GCRV 的能力。基因编辑也在农牧产品的育种中得到了广泛应用,肌肉生长抑制素(MSTN)基因对肌肉生长发育具有重要调控作用。Crispo[85]和Wang[86]两个课题组分别利用CRISPR/Cas9 技术,获得了高产肉性能的MSTN 缺失羊和表现出“双肌”表型的MSTN 双等位基因敲除猪。MSTN 的基因编辑也在团头鲂(Megalobrama amblycephala)[87]、沟鲶(Ictalurus punctatus)[88]和黄颡鱼(Pelteobagrus fulvidraco)[89]中得到应用,获得了具有双肌表型的新种质。CRISPR/Cas9系统也被广泛应用于植物领域的烟草(Nicotiana tabacum)、拟南芥(Arabidopsis thaliana)、水稻(Oryza sativa)、小麦(Triticum aestivum L.)、玉米(Zea mays)和番茄(Solanum lycopersicum)等多种植物。Shimatani 等[90]通过将Cas9 与胞苷脱氨酶(Target-AID)进行融合,再由sgRNA 进行引导,从而在水稻中获得多个除草剂抗性位点突变,并在番茄中形成了纯合可遗传的植株。Li 等[91]将nCas9 与tRNA 腺苷脱氨酶融合开发了植物腺嘌呤碱基编辑器,在水稻中进行点突变产生耐除草剂的水稻。这两项研究同样证明了CRISPR/Cas9 编辑对作物改良的可行性,扩展了植物中基因育种的工具。
5.3 CRISPR/Cas9 在动物模型构建和基因治疗中的应用
动物模型是了解人类疾病发病机制和开发新型基因治疗的有效工具。CRISPR/Cas9 具有效率高、速度快、可遗传能力强以及简单经济等优点,在动物模型和基因治疗中有着广阔发展空间。Li 等[92]通过CRISPR/Cas9 系统构建了Uhrf2 基因缺失小鼠和双基因缺失大鼠、小鼠模型。Xue 等[93]研究表明CRISPR/Cas9 可以直接用于修饰体细胞组织中的抑癌基因和癌基因,为疾病模型的快速开发提供了一种新方法。2019 年Xu[94]等利用CRISPR/Cas9 基因编辑技术,编辑修饰了人成体造血干细胞中CCR5基因,修饰后的成体造血干细胞成功重建了人体的造血系统,且未发现脱靶效应和副作用。2020 年Stadtmauer 等[95]领导的一项多重CRISPR/Cas9 编辑改造的T 细胞治疗癌症的临床试验。结果表明,该疗法在治疗癌症患者中的安全性和可行性,也为基于CRISPR/Cas9 系统在基因治疗中的作用添加了又一例证。2020 年4 月Lu[96]研究团队在《Nature Medicine》报道了使用CRISPR/Cas9 系统编辑非小细胞肺癌患者T 细胞PD-1 基因用以治疗肺癌的首个人类I 期临床试验结果,表明CRISPR/Cas9基因编辑的T 细胞的临床应用安全可行。这三项研究均在成体T 细胞上进行,并未在体内直接进行编辑修饰,因此并不会对其他组织器官及生殖系统产生影响。
6 总结与展望
迅速发展的CRISPR/Cas9 介导的基因编辑技术正在现代生命科学中产生巨大的作用。该技术可以高效、通用和简单地进行精确的基因组编辑、转录调控和表观遗传修饰。毫无疑问,CRISPR 系统具有革新基因和医学领域的潜力,最近已有CRISPR/Cas9 基因编辑技术的相关的临床试验[94,95]。然而,在CRISPR/Cas9 潜在的脱靶效应、CRISPR 组分毒性作用以及Cas9 核酸酶的免疫原性上还需要更多的科学验证。尽管可以通过不断优化改进sgRNA 长度[43,44]、使用变体Cas9(nCas9[7,33]、dCas9[49]、xCas9[56]和spCas9-NG[55])以及开发新的单碱基[65,66]和双碱基编辑器[71-73]来提高基因修饰的效率和特异性,但是还有很多基因编辑问题有待改进。因此,需深入研究CRISPR/Cas9 系统介导基因编辑的细胞内分子机制,以便其在各领域上得到适当的应用。
猜你喜欢
北方牧业(2023年13期)2023-07-28 06:50:54
生物技术通报(2023年2期)2023-03-07 12:54:58
中国实用医药(2021年20期)2021-08-04 14:27:22
无机化学学报(2020年7期)2020-07-20 02:06:44
三农资讯半月报(2020年8期)2020-05-13 14:26:35
中国医药指南(2018年3期)2018-03-23 09:14:25
中国病理生理杂志(2017年2期)2017-01-17 03:59:14
中国药业(2014年21期)2014-05-26 08:56:45
现代检验医学杂志(2014年6期)2014-02-02 03:01:54
食品工业科技(2012年1期)2012-11-15 02:03:32
