
六苯基氮杂氟硼二吡咯的合成探究和光谱测试
2022-06-27 10:00王章翠张恩立沈婧祎盛万乐
长春师范大学学报 2022年4期
王章翠,张恩立,沈婧祎,盛万乐
(蚌埠医学院,安徽 蚌埠 233030)
近红外有机共轭分子由于其具有优异的光电性质和灵活的可修饰性受到广泛关注[1-2]。例如,二吡咯甲川可以与很多过渡金属络合形成刚性平面结构,使分子荧光增强,因此可被用于开发荧光探针材料;其形成稳定的配合物也可以开发具有光电活性的功能材料,最具有代表性的就是与二氟化硼络合生成氟硼二吡咯甲川(BODIPY),该类分子在500~520 nm具有很强的发光效率,也被广泛应用于光敏剂和生物荧光成像领域[3]。将二吡咯甲川中间位置的碳用氮替换形成亚氨基桥连二吡咯(ADP),可以使化合物的光谱发生红移,对应的氮杂氟硼络合物(azaBODIPY)的吸收和发射波长达到650 nm以上,接近近红外区。
氟硼络合的二吡咯甲川和氮杂氟硼二吡咯(azaBODIPY)类荧光染料的光谱性质主要取决于配体的结构。通过在配体上引入供电子基或同时引入吸电子基团和给电子基团在分子形成推拉电子效应,都可以降低能隙,使其光谱进一步红移[4]。除此以外,在母体结构上引入并环增加分子的共轭体系也能实现光谱红移,并且和电子效应相比,引入共轭并环结构使光谱发生红移的作用更加明显。氮杂氟硼二吡咯由于其母体本身具有较长波的吸收和发射而得到广泛关注,将氮杂氟硼二吡咯并环衍生可以使其光谱发生进一步红移得到近红外材料。例如,LU等[5]利用芳基格氏试剂与邻苯二甲腈作用,开发了β,β-位苯并结构azaBODIPY,吸收发射波长红移至730~790 nm。WU等[6]从噻吩并吡咯甲酸酯出发,先合成多芳基取代的噻吩并吡咯这一重要原料,再与亚硝酸钠反应开发了α, β-位噻吩并环修饰的azaBODIPY,吸收发射波长红移至770~820 nm。在近期的工作中,SHENG等[7-8]利用金属催化和多芳基氧化偶联的方法,分别构建出β, β、α, β以及全并环的氮杂氟硼二吡咯结构,其中以全并环结构azaBODIPY为母体的化合物发射光谱的最大峰可达到920 nm。并且发现不同的并环方式也会对分子的能级结构产生很大影响。目前,氮杂氟硼二吡咯的几种不同并环结构都已经被合成出来,且都表现出优异的近红外光学性质。
利用氧化偶联的方法可以方便快捷地在azaBODIPY分子上构建并环结构。但由于azaBODIPY母体合成的限制,其修饰位点较少,结构拓展方法较为单一。如何构架氧化产物的前体分子成为关键问题。本研究先利用azaBODIPY传统的合成方法合成母体结构,经NBS溴化、Suzuki偶联得到六芳基azaBODIPY,并对Suzuki反应条件进行探究和分析,得到最优的合成条件。合成路线见图1。
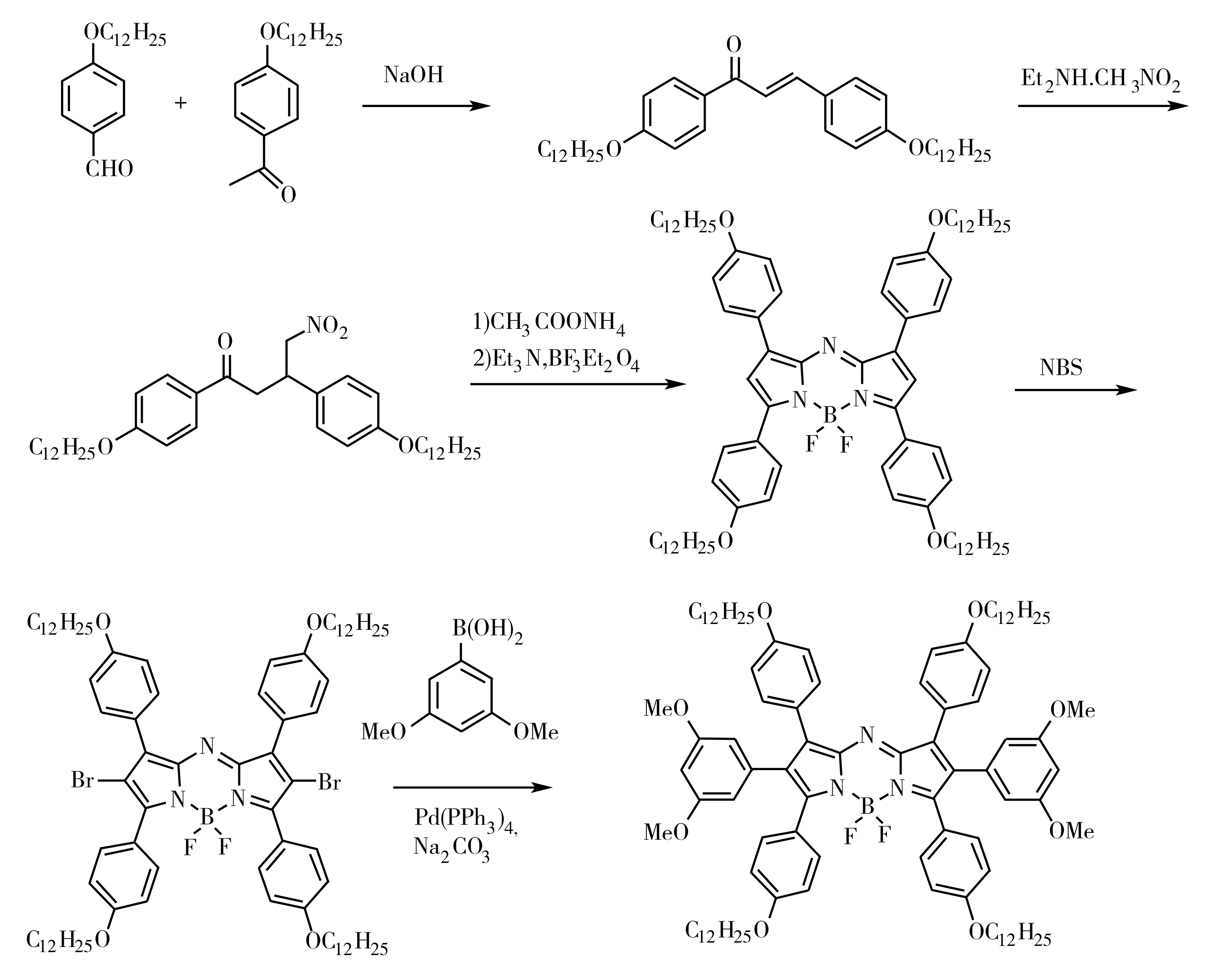
图1 六苯基氮杂氟硼二吡咯的合成路线
1 实验部分
1.1 仪器与试剂
电子天平(FA1004,天津天马恒基仪器有限公司)、磁力搅拌器(DF-101S,河南兄弟仪器设备有限公司)、油浴锅(甲基硅油)、核磁共振(Bruker AVANCE 300MHz)、分光光度计(UV-2450)、荧光光谱仪(Edinburgh FlS-920)。
对十二烷氧基苯甲醛、对十二烷氧基苯乙酮、氢氧化钠、二乙胺、硝基甲烷、醋酸铵、NBS、四三苯基膦钯、碳酸钠、乙醇、甲苯、二氯甲烷、石油醚。所有试剂和溶剂若无特殊说明均为试剂公司购买(分析纯)直接使用没有进一步纯化。
1.2 化合物的合成
1.2.1 长链azaBODIPY的合成
将乙醇60 mL、对十二烷氧基苯甲醛(5.0 g,0.017 mol)、对十二烷氧基苯乙酮(5.2 g, 0.017 mol)依次加入250 mL圆底烧瓶中搅拌,待上述反应物溶解后加入氢氧化钠(1.7 g,0.043 mol)。持续搅拌10 h,反应析出大量沉淀,减压过滤得粗产品8.7 g,粗产率89%。粗产品不经过进一步纯化直接进行下一步反应。
将上步反应得到的产物,六倍当量的二乙胺、硝基甲烷以及乙醇100 mL,加入至250 mL圆底烧瓶中,加热回流12 h。待原料反应完全后,减压蒸馏除去溶剂和剩余二乙胺和硝基甲烷。向反应体系中重新加入乙醇溶剂100 mL和醋酸铵15 g,继续回流24 h,减压过滤得黑色蜡状固体3.0 g。再将黑色固体加入到圆底烧瓶中,加入甲苯50 mL溶解,油浴加热至60℃,加入三乙胺5 mL和三氟化硼乙醚5 mL,反应2 h,利用柱层析分离得到azaBODIPY 2.1 g,总产率23%。
1.2.2 二溴化azaBODIPY的合成
称取azaBODIPY (1.80 g, 1.5 mmol)加入250 mL圆底烧瓶内,加入80 mL二氯甲烷溶解,冰浴条件下,边搅拌边滴入NBS的二氯甲烷溶液(0.55 g, 3.1 mmol),滴加完成后继续反应0.5 h,反应完全后利用柱层析除去杂质。减压蒸馏除去溶剂,得黑色固体1.90 g,产率为92%。
1.2.3 六芳基azaBODIPY的合成
称取上步得到的二溴化azaBODIPY(0.50 g, 0.36 mmol)、四三苯基膦钯(0.05 g, 0.03 mmol)、3,5-二甲氧基苯硼酸(0.26 g, 1.44 mmol)加入反应器中,再加入甲苯40 mL和饱和碳酸钠溶液20 mL,在氮气保护下,90℃搅拌反应24 h,反应结束后,取有机相干燥处理,利用柱层析分离得暗红色金属光泽固体0.48 g,产率88%。
目标化合物的核磁谱图:1H NMR (300 MHz, CDCl3) δ 7.50-7.41 (m, 8H), 6.77-6.76 (m, 8H), 6.32 (s, 2H), 6.17 (s, 4H), 4.05-3.84 (m, 8H), 3.57 (s, 12H), 1.89-1.68 (m, 8H), 1.52-1.12 (m, 72H), 0.88 (t,J= 6.5 Hz, 12H)。13C NMR (75 MHz, CDCl3) δ 160.5, 160.4, 159.5, 157.8, 145.1, 140.3, 135.5, 132.5, 132.5, 132.2, 131.8, 124.3, 122.9, 113.8, 113.7, 108.6, 99.9, 68.0, 67.9, 55.2, 31.9, 29.6, 29.4, 29.3, 29.2, 26.1, 26.0, 22.7, 14.1。
2 结果与讨论
2.1 反应条件对Suzuki反应产率的影响
本研究考察了反应条件,如反应温度、催化剂用量、反应时间对Suzuki偶联反应最终产物产率的影响(表1)。固定其它反应条件不变,发现在催化剂为10%、温度为90℃下反应24 h,产率可达88%~89%。可能是因为卤素两边连接芳香环的影响,使得催化反应具有很大位阻,反应速率较慢。如果反应时间少于24 h,就会发现反应物有剩余。不仅如此,反应也需要较高温度,反应低于90℃时,反应24 h后依旧会有原料剩余。催化剂为10%时效果最佳,低于10%可能需要更长的反应时间,高于10%催化剂的用量增加也不能明显提高产率,还会造成试剂的浪费。

表1 反应温度、反应时间和催化剂用量对Suzuki反应的影响
2.2 化合物的吸收和发射光谱
本研究测试了化合物六芳基氮杂氟硼二吡咯的吸收光谱(图2)和发射光谱(图3)。在二氯甲烷中,化合物的最大吸收波长为675 nm,最大发射波长为717 nm。虽然增加两个苯环,但和四芳基azaBODIPY母体化合物相比并没有明显的光谱红移,可能是因为引入的苯环增加芳环之间的位阻,使外围苯环与母核平面之间具有较大的二面角,单个苯环对母体的供电子能力有所减弱。

图2 六苯基氮杂氟硼二吡咯在二氯甲烷中的归一化吸收谱图
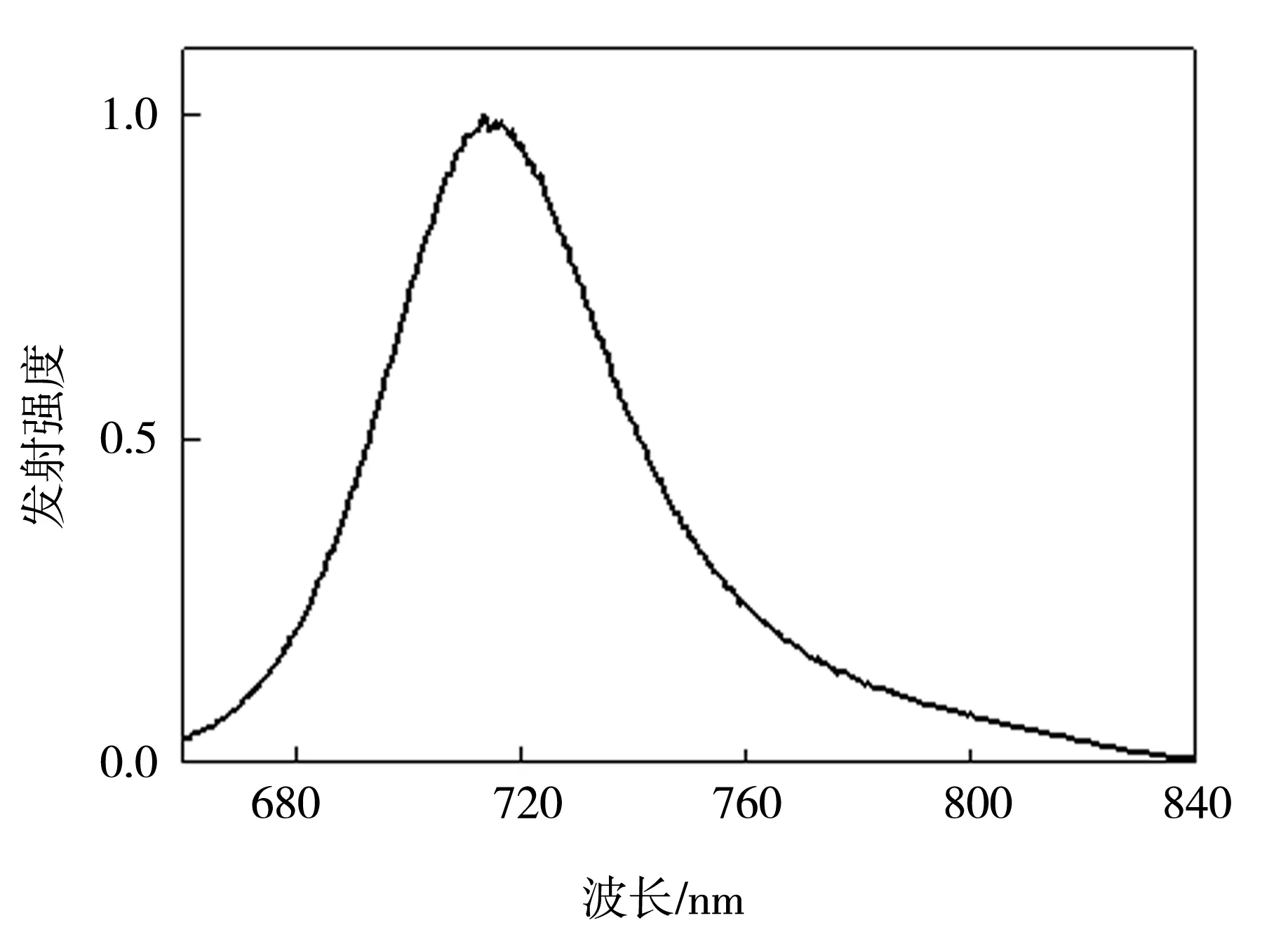
图3 六苯基氮杂氟硼二吡咯在二氯甲烷中的归一化发射谱图
3 结语
本文以对十二烷氧基苯甲醛、对十二烷氧基苯乙酮和芳基硼酸等为原料,经过羟醛缩合、迈克尔加成、成环和氟硼络合四步反应合成了具有长链的四芳基氮杂氟硼二吡咯,再经过溴化反应、Suzuki偶联得到六芳基氮杂氟硼二吡咯。同时,还对Suzuki反应的条件进行尝试,探究出最优的反应条件。最后对所合成的产物进行了光谱测试,得知其最大吸收波长为675 nm,发射波长为717 nm。
猜你喜欢
中南民族大学学报(自然科学版)(2022年6期)2022-11-02
分子催化(2022年1期)2022-11-02
化学工程师(2022年3期)2022-04-19
四川轻化工大学学报(自然科学版)(2021年3期)2021-08-30
上海化工(2021年2期)2021-04-23
中南民族大学学报(自然科学版)(2020年6期)2020-12-22
化学与粘合(2020年4期)2020-09-11
生物工程学报(2020年1期)2020-03-12
科技与企业(2015年20期)2015-10-21
中国塑料(2015年10期)2015-10-14
