
聚乙烯醇/相变微胶囊多孔定形复合相变材料的制备及性能
2022-06-22 08:26:26张曼妍师文钊刘瑾姝陆少锋崔杉杉周红娟苏国鑫
高分子材料科学与工程 2022年2期
张曼妍,师文钊,刘瑾姝,陆少锋,崔杉杉,周红娟,苏国鑫
(西安工程大学纺织科学与工程学院,陕西 西安 710048)
近年来,能源的消耗以及环境污染成为社会广泛关注的问题。由于太阳能等可再生能源存在间歇性、波动性等缺点,能够实现空间和时间的自由调度的能源即储能技术引起了广泛研究[1]。其中相变储热材料由于其蓄热密度与蓄热容量大,成本低等优点[2],被广泛应用于建筑[3]、航空航天[4]、太阳能储热[5]等领域。相变材料根据相变状态通常分为固液相变材料、固固相变材料和液气相变材料[6],其中,固液相变材料具有潜热密度高、成本低、选择性强等优势,然而这类相变材料在相变过程中常出现液体泄漏,导致储能效率降低及环境污染的问题。针对这些问题,可以使用多种方法以减少泄漏,提高相变材料利用率。
相变复合材料是定形相变材料中的一种类型,通常是通过将相变材料嵌入到有机或无机支撑材料中制得[7]。多孔材料具有比表面积大、导热率高等优点,被广泛用于相变材料的定形封装,从而解决固液相变材料泄露等问题[8]。聚乙烯醇(PVA)是一种多羟基强极性高分子材料,具有良好的耐热性能、耐酸碱稳定性及生物相容性[9],同时侧链含有大量羟基,易发生交联。作为定形材料,多孔结构的PVA 常用于相变复合材料的制备[10]。此外PVA 分子链经过化学交联或物理结晶区形成三维网络可以作为固定相,无定形区作为可逆相,使其具有形状记忆功能[11];由于PVA 易于与水结合,因此具有湿致型形状记忆性能。
然而目前制备PVA 基多孔材料常使用淀粉作为成孔剂,成孔后需消耗大量水来洗除,生产过程中存在很大的浪费和污染。因此本文以PVA 多孔材料为基材,采用物理发泡法将微胶囊封装的相变材料与多孔材料复合赋予复合材料相变性能与湿热双重响应形状记忆性能。
1 实验部分
1.1 试剂与原料
聚乙烯醇:聚合度1750±50,无锡市亚泰联合化工有限公司;β-环糊精:孟州市华兴生物化工有限责任公司;氯化镁:分析纯,天津市百世化工有限公司;树脂整理剂EFR-3:深圳先进华联精细化工有限公司;微胶囊乳液:以异氟尔酮二异氰酸酯(IPDI)与二乙烯三胺(EDTA)为反应单体、硬脂酸丁酯为相变材料,通过界面聚合法制得,相变温度20 ℃,潜热焓值(86.5±5) J/g,粒径20μm[12];柠檬酸:分析纯,天津博迪化工有限公司。
1.2 多孔定形复合相变材料的制备
将PVA 粉末于95 ℃加热溶解配制质量分数为15 %的均匀溶液。将2%柠檬酸、2 %氯化镁、14 %EFR-3 树脂及8 %β-环糊精加入PVA 溶液中共混均匀,再将相对于PVA 质量10%,20%,40%,60%的相变微胶囊乳液分别加入上述溶液中得到共混发泡溶液。搅拌均匀后以1000 r/min 的转速搅拌10 min,物理发泡形成稳定的泡沫体系后倒入模具中,置于70 ℃烘箱中固化交联烘干5 h,即得到PVA/相变微胶囊多孔定形复合相变材料。样品分别标记为PCMs-10,PCMs-20,PCMs-40,PCMs-60,未添加相变微胶囊乳液的样品标记为PCMs-0,样品制备过程如Fig.1 所示。
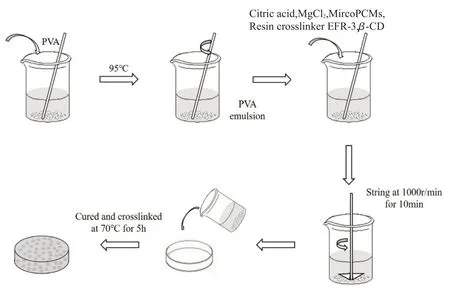
Fig.1 Preparation process of the sample
1.3 测试与表征
1.3.1 表观密度测试:将制得的材料裁剪为规则的长方体,测量其长宽高后计算体积(V),在精度为0.001 g 的天平上测量其质量(m),计算其表观密度(ρ)。
1.3.2 扫描电镜分析:裁取较平整的样品于真空中喷金后进行扫描拍照。采用场发射扫描电子显微镜(SEM,美国FEI 公司,Quanta-450-FEG)观察材料的孔隙结构。
1.3.3 光学显微镜分析:将物理发泡后的溶液取少量置于载玻片上,使用光学显微镜(XTL-5W)观察其泡孔形态。放置30 min,1 h 后再次观察其形态。
1.3.4 泡孔稳定性测试:采用2 种方法对发泡后的溶液泡孔稳定性进行测定。将发泡后的溶液倒入试管中记录其高度(h0),放置30 min 后的液面高度记录为(h1),根据式(1)计算其气泡逸散率(R)。同时使用高级旋转流变仪(奥地利安东帕,MCR302)测定其黏度。

将已吸附平衡的样品取出,用滤纸擦去表面水分后称量(mi,单位g),在离心管中塞入一些滤纸,将样品放入离心管中,使用高速离心机(TDL-40B,上海安亨科学仪器厂)以1000 r/min 的转速离心5 min,取出样品称量其质量(mr,单位g),按照式(4)计算保水率(R)。

1.3.7 相变性质分析:使用差示扫描量热仪(瑞士METTLER,DSC-1)进行熔融潜热(ΔHm)的测试。温度设定为-20~70 ℃,升温及降温速率为10 ℃/min,保护气氛为氮气,流量为50 mL/min。
1.3.8 热重分析:使用热重同步分析仪(瑞士METTLER,TGA2)进行热重测试。温度设定为30~600 ℃,升温速率为10 ℃/min,保护气氛为氮气,流量为50 mL/min。
1.3.9 形状记忆性测试
(1)热致形状记忆性能:取一块去除表面膜的样品,在60 ℃烘干2 h 使其固定为90°折角,取出冷却至室温记录其角度(λ2)。放入80 ℃的烘箱中并计时,记录规定时间后的角度(λ0),分别根据式(5)、式(6)计算其形状固定率(Rf)与热诱导形状记忆回复率(Rr)。
(2)湿致形状记忆性能:将样品固定为90°折角在60 ℃的烘箱中烘干2 h,使其产生形状变化后取出,使用滴管在样品褶皱的部分滴水,计算其形状固定率(Rf)与形状回复率(Rr)。计算公式为式(5)、式(6),形状记忆原理见Fig.2。
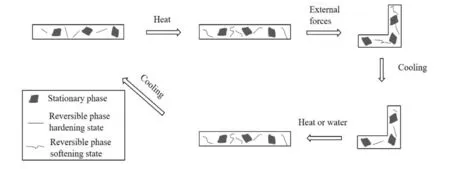
Fig.2 Principle of the shape memory

2 结果与讨论
2.1 密度分析
所制备不同PVA/相变微胶囊多孔定形复合相变材料样品密度测试结果如Tab.1 所示。由Tab.1 可以看出,加入微胶囊乳液的样品密度均比不加入微胶囊乳液的样品大,且随着微胶囊乳液用量的提高,样品表观密度减小,但相差不大。这是因为复合微胶囊乳液的样品中含有微胶囊及乳化剂等,而随着微胶囊乳液用量的提升,小泡孔形态不稳定而出现并孔现象,孔径变大,样品结构越来越疏松,因此样品密度随微胶囊乳液用量提升而降低。

Tab.1 Density of samples
2.2 孔结构分析
不同样品的SEM 照片如Fig.3 所示。可以看出,所得材料泡孔结构完整且较为规整。由于材料以物理搅拌法成型,存在气泡逸散、并泡等情况,因此兼有闭孔和开孔结构,即泡孔呈闭孔状态,孔壁上出现较多小孔。随微胶囊用量增大,大小孔孔径逐渐增大。大孔平均孔径分别为403.4μm、542.6μm、548.8μm、552.3μm 和573.4μm,小孔平均孔径分别为99.4μm、173.8μm、174.0μm、180.5μm和204.2μm。这是因为,随微胶囊乳液用量增加,乳化剂含量随之增加,所制备的原液稠度下降,在搅拌和加热成型过程中泡孔产生逸散,稳泡效果下降,出现并泡现象[13],导致材料孔径增大,均匀性下降。

Fig.3 SEM images of PCMs-0(a),PCMs-10(b),PCMs-20(c),PCMs-40(d)and PCMs-60(e)
为进一步研究泡孔形成,利用光学显微镜(放大倍数为400 倍)观察了刚发泡、放置30 min 及放置1 h 后的复合乳液,如Fig.4 所示。刚发泡的乳液中,出现完整、大小不一的球形泡,泡孔壁可见少量小泡;放置30 min 后,球形泡壁上小泡明显增多,接触并结合,泡壁表面形成凸起状;放置1 h 后,合并小泡增多,泡孔壁上小泡尺寸增加,与SEM 图相符合。出现这种泡孔结构是由于搅拌发泡过程中,泡孔逐渐产生,直径不均一,逸散速率不同,在逸散过程中出现了泡孔之间的接触与结合,使其产生了大孔表面有小孔相互连通的现象。不同微胶囊含量的复合乳液的黏度与气泡逸散率如Tab.2 所示,随着微胶囊乳液用量的增加,溶液的黏度逐渐下降,放置30 min 后的气泡逸散率逐渐增加,这与黏度结果相符合。这是由于微胶囊用量的提高会降低混合溶液体系的泡孔稳定性,使其更易发生逸散与并泡现象,最终造成多孔定形复合相变材料的孔结构发生变化。

Tab.2 Viscosity and bubble dissipation rate of samples

Fig.4 Optical microscope images of foamed emulsions after 0 min(a),30 min(b)and 1 h(c)
2.3 力学性能
不同微胶囊乳液含量的复合材料的拉伸强度如Fig.5 所示。由图可见,随微胶囊乳液含量增加,复合材料拉伸强度先增加后降低。这与微胶囊乳液含量变化导致的泡孔结构变化有关。适量微胶囊乳液的加入,有利于形成完整且均一的泡孔,获得较好的力学强度;但过多微胶囊乳液加入,复合材料泡孔均匀性下降,孔径增大,孔壁变薄并破裂,抵抗外力变形能力减小,易发生断裂,力学强度下降。

Fig.5 Tensile stress of samples
2.4 吸水和保水性
PVA/相变微胶囊多孔定形复合相变材料的吸水率随浸泡时间的变化曲线如Fig.6 所示。由图可得,复合材料浸入水中后,吸水率快速上升,并迅速达到吸附平衡,说明吸水性良好。这是因为PVA 链段上含有大量羟基,与水分子间可形成强的分子间作用力。此外,由于存在一定的开孔,相对闭孔材料,相互连通的三维网状孔结构更有利于水分子快速进入样品内部并被吸附,使其具有良好的吸水性。
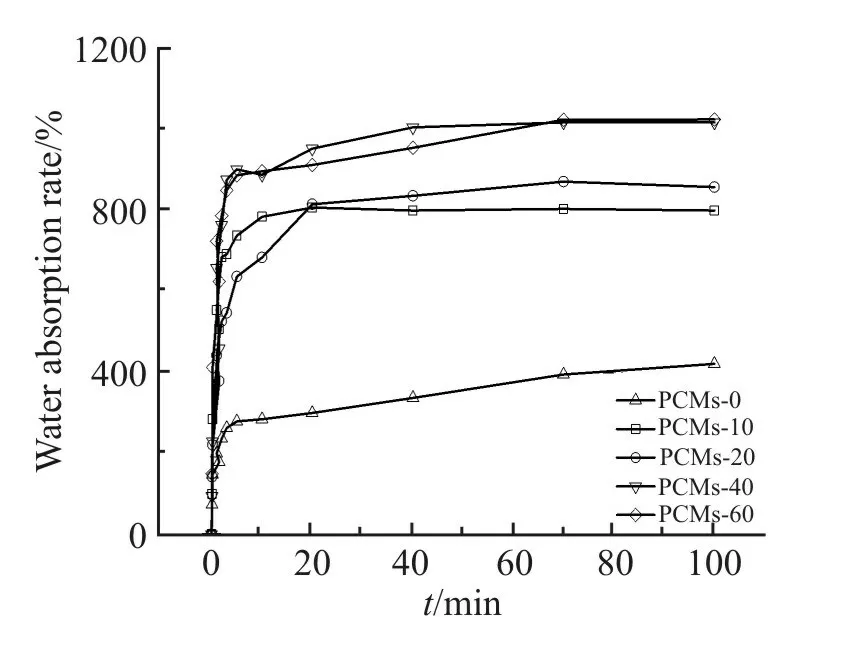
Fig.6 Variation of water absorption rate of samples vs.time
PVA/相变微胶囊多孔定形复合相变材料样品的平衡吸水率见Tab.3。从Tab.3 可得,PVA 多孔复合材料的平衡吸水率随着微胶囊乳液用量的增加,从426.3%上升到1025%,增幅明显。这是因为随着微胶囊质量比的增加,微胶囊乳液中乳化剂用量提高,使复合乳液的黏度降低,流动性增强,物理发泡过程中,泡孔易发生逸散并泡,成型后材料泡孔尺寸较大,密度降低,更容易吸附水分。

Tab.3 Water absorption rate of samples
样品保水率如Tab.4 所示。可以看出,随着微胶囊乳液用量的增加,样品的保水率呈现下降趋势,但整体保水率仍高于未加微胶囊乳液的样品。这是因为微胶囊乳液用量的增加使物理发泡所得泡沫体系流动性增强,最终得到样品的孔隙结构更加疏松,泡孔尺寸越大,水分子可快速进入样品内部并与PVA 羟基形成更多氢键,即使在离心力作用下脱附,残留量仍较大。但泡孔孔径增大,水分子可通过的孔道增大,在离心作用下更易发生脱附,因此样品保水率随着微胶囊用量的提高反而降低。

Tab.4 Water retention rate of samples
2.5 相变性能
Fig.7(a)与Fig.7(b)分别是样品升温与冷却的DSC 曲线。从图中可以看出,复合相变材料具有相变性能,相变温度(Tm)为20℃,相变微胶囊的用量对材料的相变温度基本不产生影响。其中,冷却时出现2 个结晶峰,这是烷基PCMs 由α固相向β固相发生转变所致[14]。各样品的潜热焓值如Tab.5 所示,可见,随着相变微胶囊含量的增加,材料的潜热焓值也随之增加,焓值最大可达到10.85 J/g,具有良好相变性能。

Tab.5 ΔH of samples

Fig.7 DSC(a)heating and(b)cooling curves of samples
此外,由于微胶囊纳米颗粒具有表面积大、表面有大量电荷以及颗粒间有较强氢键等特征,易团聚,导致复合材料出现相变性能不均匀现象。为了验证制得的复合材料中微胶囊的均匀性,随机取样,裁取3 个不同部位进行了测试。经测试可知,所有样品偏差不超过2.6%,表明所制备的PVA 相变复合多孔材料中微胶囊纳米颗粒分散均匀。
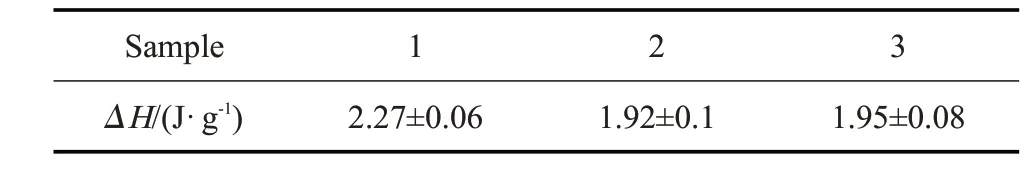
Tab.6 ΔH of different parts of samples
2.6 热稳定性
Fig.8 为PVA/相变微胶囊多孔定形复合相变材料样品的TG 曲线。由Fig.8 可得,所有样品的热分解曲线基本一致,主要分为3 个阶段,最大分解速率出现在200 ℃左右。 30~150 ℃,曲线基本保持不变,有极少量的质量损失,源于复合材料中自由水与结合水的蒸发;200~450 ℃,PVA 很快分解,生成醋酸、乙醛、丁烯醇和水,曲线质量损失变化较大,质量损失率在450 ℃左右达到最大;450~600 ℃,发泡材料主体已经炭化,复合材料质量变化不大。

Fig.8 TGA curves of samples
材料的最大热分解温度及质量残余率如Tab.7所示。由表可知,在升温速率为10 ℃/min 的条件下,不同样品的最大热分解温度相差不大,最多不超过8 ℃,说明PCMs 的加入对材料耐热性能影响不大,这是由于微胶囊乳液与PVA 之间没有化学键与共价键的结合,而是通过氢键、范德华力等次价键相结合的。且加入微胶囊乳液的复合材料质量残余率均较未加入微胶囊乳液的低,说明相变微胶囊在高温下分解得更为彻底。
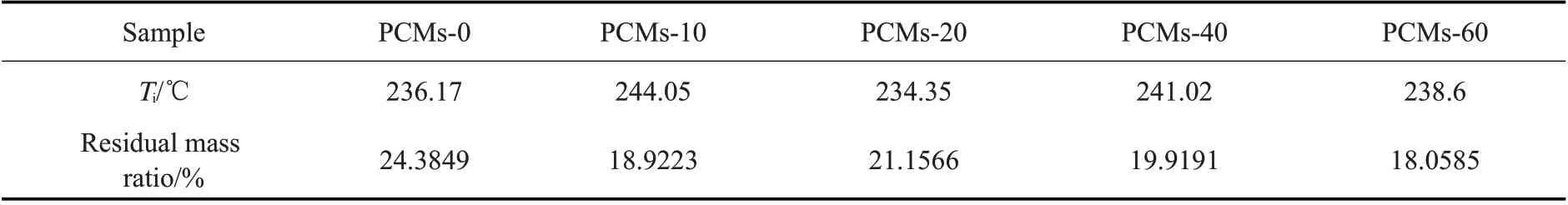
Tab.7 Ti and residual mass ratio of samples
2.7 形状记忆性能
PVA 基多孔定形复合相变材料形状记忆性能结果如Tab.8 所示。热诱导与湿诱导后,材料均有较明显的回复现象;同时可以看出,湿诱导后材料的回复较热诱导后的回复率更大。这是因为随着水分引入,材料发生吸湿溶胀,水分进入复合材料的无定型区,破坏变形态内部氢键,使材料在固定相作用下更快回复到原始形态。微胶囊乳液用量与其形状记忆性能没有明显关系,这可能是因为PVA复合材料的形状记忆特性取决于PVA 链段的结晶与熔融,微胶囊的共混未明显改变PVA 分子链相互作用。但加入40%与60%微胶囊乳液的复合材料样品的形状回复率较大,这是由于加入大量乳液的样品结构松散,导热与吸水性能较好,在相同时间内外界影响因素更容易刺激样品内部大分子链相互作用,破坏变形态中形成的新键从而发生形状回复。

Tab.8 Rf and Rr of samples
3 结论
采用物理发泡法与溶液浇筑法将相变微胶囊与PVA 溶液复合后成功制备得到一种兼具相变性能与形状记忆性能的复合材料。复合材料呈稳定的发泡材料状态,材料孔结构以闭孔结构为主,存在部分开孔结构,具有较小的密度,断裂强力可达到33.20 kPa,此外复合材料保留了PVA 本身的特性,具有良好的吸水性与保水性,吸水率可达到1025%。复合材料具有相变储热性能与良好的形状记忆性,热焓值最大可达到10.85 J/g, 在热刺激和水刺激下均有较大的形状回复,形状回复率可以达到83.3%,具有良好的湿热双重响应形状记忆性能。通过改进发泡与成型方法将相变微胶囊与多孔材料复合制得的材料减少了环境污染,并赋予其相变性能,有望在抗皱织物材料以及电子元件过热保护装置等方面起到作用。
猜你喜欢
初中生学习指导·中考版(2022年1期)2022-02-09 11:46:09
包装工程(2022年1期)2022-01-26 09:03:10
科教导刊·电子版(2021年6期)2021-05-06 05:05:14
工程塑料应用(2020年11期)2020-11-28 01:57:50
中国塑料(2016年10期)2016-06-27 06:35:12
中国塑料(2016年8期)2016-06-27 06:34:44
材料科学与工程学报(2016年5期)2016-02-27 07:11:29
合成材料老化与应用(2015年4期)2015-07-25 10:45:44
河南科技(2015年1期)2015-02-27 14:20:19
兴趣英语(2013年6期)2013-08-29 07:45:26
