
Li7P3S11电解质的合成、传导及应用
2022-06-14 09:03彭林峰李莉萍程时杰
无机化学学报 2022年6期
廖 聪 余 创 彭林峰 李莉萍 程时杰 谢 佳
(1华中科技大学电气与电子工程学院,强电磁工程与新技术国家重点实验室,武汉 430074)
(2华中科技大学材料科学与工程学院,武汉 430074)
(3吉林大学无机合成与制备化学国家重点实验室,长春 130012)
0 引 言
锂离子电池因其能量/功率密度和成本优势,在便携电子设备、电动汽车、大规模静态储能等领域应用广泛[1-3]。随着锂离子电池商业化规模的不断扩大,使用液态有机电解液的锂离子电池的问题逐渐暴露出来,如安全性低、能量密度低等[4-7]。采用不易燃的固态电解质替换易燃的液态有机电解液[3,8-9],不仅提高了电池的安全性,还能减少电池中非活性物质的使用;此外,固态电解质的引入使得金属锂负极的应用成为可能[6],提升了锂离子电池的能量密度。将液态有机电解液替换为固态电解质构筑而成的全固态电池,有望取代传统锂离子电池,成为下一代储能技术。
在固态电池中,固态电解质不仅需要传导Li+,而且需要充当隔膜避免正/负极材料直接接触。因此,对于固态电解质的理化性质要求包括[10-11]:(1)在室温下具有高离子电导率(>10-3S·cm-1);(2)宽电化学窗口(>5.0 V(vs Li/Li+));(3)低电子电导率(10-8S·cm-1);(4)对正/负极材料具有化学稳定性;(5)离子迁移数约为1;(6)电解质与电极活性材料的烧结温度匹配;(7)低毒性和低成本。常见的固态电解质可以分为氧化物[12-14]、硫化物[15-17]、聚合物固态电解质[18-19]三类,此外还有氮化物[20]、卤化物[21]、磷酸盐[4]等固态电解质。聚合物虽然具有较好的力学性能,但室温电导率很低。与之相比,硫化物固态电解质电导率高于聚合物、氧化物固态电解质[22],力学性能也更加优越[11]。硫化物固态电解质是通过S原子取代氧化物固态电解质中O原子演变而来,其较高的离子电导率主要来自以下2个因素:(1)S2-的半径大于O2-,使得硫化物能够提供更大的Li+传导通道;(2)S原子电负性小于O原子,使得S2-与Li+之间相互作用小于O2-与Li+[23-24]。固态电解质的离子电导率对于全固态电池的性能有重要影响,因此开发高电导率的硫化物固态电解质对于构筑高性能全固态电池至关重要。
近年来,关于硫化物固态电解质材料的研究不断深入。其中,具有较高离子电导率的Li10GeP2S12(1.2×10-2S·cm-1)[25]和 Li7P3S11[26](1.7×10-2S·cm-1)两种硫化物固态电解质材料引起学者广泛关注。合成Li10GeP2S12的原材料含有成本价高的Ge元素,阻碍了其在固态电池中的规模化应用;与之相比,Li7P3S11的原材料价格相对较低廉、电导率亦具有可比性等优势,有助于其在固态电池领域实现实用化。尽管具有上述优点,Li7P3S11电解质的实用化进程仍面临诸多问题,如对空气/水稳定性差[23]、与电极界面稳定性差[27-28]、电导率与有机液态电解质仍存在较大差距[29]、化学/电化学稳定性差[17]等。
本文主要综述了关于Li7P3S11固态电解质的合成路径、晶体结构及锂离子传导机理、离子电导率改善、稳定性改善及全电池的应用等各个方面的研究进展。详细探究了Li7P3S11电解质的结构和传导/工作机理等基础研究,总结了针对Li7P3S11理化性能改善所采取的各种策略,评述了Li7P3S11在全固态锂离子电池和全固态锂硫电池中的应用。最后,展望了Li7P3S11固态电解质的未来研究方向。
1 Li7P3S11电解质的结构和锂离子传导机理
1.1 Li7P3S11电解质的结构
Li7P3S11的晶体结构最早由Tatsumisago等[30]通过同步辐射X射线衍射并对测试结果采用全局空间优化和Rietveld精修而得到。如图1a[31]所示,Li7P3S11晶体结构属于三斜晶系,空间群P,每个晶胞具有2个Li7P3S11单元。如图1b[30]所示,其晶体结构可以看作由[PS4]3-四面体和[P2S7]4-双四面体构成。根据晶体对称性,Li、P、S三种原子分别占据7、3、10种位置;Li+主要分布在[PS4]3-四面体和[P2S7]4-双四面体的间隙位置,从Li+角度[32]而言,Li7P3S11的晶体结构也可以看作由[LiS4]7-和[S4]8-四面体通过共用顶点、棱边形成。Ceder等[33]将Li7P3S11晶体结构看作由[LiS4]7-和[S4]8-两种四面体构建而成,证明了Li7P3S11中由S2-构成的四面体呈面心立方堆积,有利于Li+在结构中快速传导。
1.2 Li7P3S11电解质的离子传导机理
Li7P3S11电解质因其高电导率获得广泛关注,而了解其高电导率背后的Li+传导机理有助于对其性能进一步改善。目前主要采用X射线衍射、中子衍射等结构表征手段并结合理论模拟计算研究Li7P3S11电解质中Li+传导机理。
早在2011年,Holzwarth等[34]通过X射线衍射、中子衍射解析了Li7P3S11电解质晶体结构,并结合第一性原理计算,提出Li7P3S11电解质结构中可存在的3种传导机理:空位机制、间隙机制、空位-间隙机制。他们采用微动弹性带法(nudged elastic band,NEB)计算了Li+在缺陷位点间的迁移能,如图1c所示,发现在空位机制中Li+沿晶体b轴在[PS4]3-四面体和[P2S7]4-双四面体之间传导的迁移能Em仅有0.15 eV。该研究对比了模拟计算获得的活化能与实验测得的实际Li+活化能,发现两者基本吻合。对于空位与间隙传导机制,Cho等[35]通过第一性原理计算发现Li+迁移能在间隙机制下的数值远低于空位机制,且间隙机制下的最小迁移能为0.24 eV。

图1 (a)Li7P3S11晶胞图[31];(b)Li7P3S11沿[010]晶向的结构图[30];(c)Li+在4和3位点沿着b轴移动的路径示意图[34];(d~f)Li+在bcc、fcc、hcp阴离子堆积方式中的传导路径(左侧)和其在S2-晶格的bcc、fcc、hcp堆积方式中传导路径的能量(右侧)[33]Fig.1 (a)Unit cell of Li7P3S11[31];(b)Structure of Li7P3S11viewed along the[010]direction[30];(c)Visualization of the path between the 4 and 3 sites having net migration along the b-axis based on the perfect crystal structure[34];(d-f)Li+migration pathways in bcc,fcc,hcp-type anion lattices(left panels)and calculated energypath(right panels)in the bcc,fcc,and hcp stacking of sulphur lattices[33]
无论上述哪种传导机制,在晶体结构中均以[PS4]3-四面体和[P2S7]4-双四面体2种基团为基础提出。此外,也有学者提出了以S2-堆积构造为基础的理论模型。Onodera等[32]基于飞行时间中子衍射和同步辐射X射线衍射结构分析结果,结合反向蒙特卡罗模型(reverse monte carlo modeling,RMC),建立了Li7P3S11玻璃-陶瓷态和玻璃态中Li+的三维传导网络结构。在该研究中提出了ac-[S4]8-(可接受Li+形成[LiS4]7-)和[S4]8-(不可接受 Li+形成[LiS4]7-)单元结构,在Li7P3S11玻璃-陶瓷态中[LiS4]7-单元的ac-[S4]8-配体数目为3.9,高于Li7P3S11玻璃态的1.9,因此前者具有更高的锂离子电导率。然而,关于S2-的堆积结构对Li+的快速传导的影响并未阐述。Ceder等[33]发现在锂离子固态电解质中Li+的快速传导与结构中阴离子的堆积方式有关,如图1d~1f中S2-的不同堆积结构所示;当 S2-以bcc方式(图 1d)堆积时,Li+沿着 2 个共面的[S4]8-四面体传导,传导路径为T-T,Li+的迁移能为0.15 eV;当S2-以fcc方式(图1e)堆积时,Li+在2个[S4]8-四面体间传导需要经过共面的[S6]12-八面体,传导路径为T-O-T,由于存在[S6]12-八面体的阻碍作用,Li+的迁移能为 0.39 eV;当 S2-以hcp方式(图 1f)堆积时,存在3种传导路径T-O-T、T-T、O-O,3种路径的迁移能分别为0.39、0.20、0.19 eV。然而,具有高离子电导率的Li7P3S11和Li10GeP2S12两种电解质中S2-均以bcc方式堆积,因此硫化物固态电解质中的S2-以bcc方式堆积时,能够形成Li+的快速传导通道。
2 Li7P3S11电解质合成路径
Li7P3S11电解质具有较高的室温离子电导率,使得它成为一种极具潜力的固态电解质。因此,探索一种低成本、可大规模制备的高性能Li7P3S11固态电解质的合成路径尤为重要。目前,该电解质的合成方法主要有3种:熔融萃取法、机械球磨法、液相合成法。研究发现晶态Li7P3S11在室温下并不能稳定存在,Ong等[36]通过计算与实验相结合,发现Li7P3S11在553 K(280℃)可以结晶化,因此在室温下,通常以无定形的玻璃态或者部分晶化的玻璃-陶瓷态存在。玻璃-陶瓷态Li7P3S11一般无法一步合成,主要是通过机械球磨结合高温热处理得到玻璃态Li7P3S11,冷却后形成部分结晶的Li7P3S11沉淀在玻璃相中,晶相和非晶相共混的组成即为玻璃-陶瓷态Li7P3S11。
2.1 熔融-萃取法
熔融-萃取法是一种简单快速制备Li7P3S11固态电解质的方法,主要是通过将原料按照化学计量比配制,然后放入抽真空的石英管中高温加热一段时间,随后采用冰水低温萃取,形成玻璃态Li7P3S11;最后,将获得的玻璃态Li7P3S11加热到结晶温度,冷却后在玻璃态Li7P3S11中形成部分结晶态,即玻璃-陶瓷态。Hayashi等[37]在2007年,将物质的量之比为7∶3的Li2S和P2S5混合后,放入具有碳涂层的石英管中,在真空中密封石英管并以不同的温度和时间熔融-萃取玻璃态Li7P3S11,然后加热制备玻璃-陶瓷态Li7P3S11,并且将其与球磨法制备的Li7P3S11电解质进行对比。通过对比XRD图发现,熔融温度为750℃时,2种方法制备的电解质具有几乎相同的晶相和结晶度;熔融温度为820和900℃时,拉曼谱图中能够明显观察到[P2S6]2-基团的存在,表明Li7P3S11已经部分分解为Li4P2S6。Holzwarth等[38]也从热力学角度分析发现,Li7P3S11在高温下容易分解为更稳定且离子电导率更低的Li3PS4和Li4P2S6两种硫化物[39]。因此,对于熔融-萃取法制备Li7P3S11电解质,控制热处理温度显得尤为重要。
2.2 机械球磨法
机械球磨法是将原材料按一定比例放入球磨罐中,并且加入一定质量的球磨珠,然后在控制球磨转速和时间的条件下,进行固相反应的过程。机械球磨法通过球磨珠的高速旋转使原材料之间发生高能量的碰撞,因此又称为高能球磨,主要包括混料、破碎、非晶化和固相反应4个过程[16]。与熔融-萃取法相比较,高能球磨具有处理温度低、杂质少的优点,是目前制备Li7P3S11电解质的主要方法。然而高能球磨制备的材料通常是玻璃态,必须要对制备的材料进行后续的热处理,才能得到玻璃-陶瓷态Li7P3S11电解质。
机械球磨法涉及的可控变量比较多,并且这些变量对合成的材料影响较大,如球磨珠和球磨罐的材质、球磨珠和原始材料的质量比、球磨时间和转速等[31];控制不同的球磨参数使得固相反应过程获得的能量不同。针对球磨罐和球磨珠材质的研究,Tatsumisago等[40]对比了Al2O3和ZrO2材质的球磨罐和球磨珠对合成电解质的影响。通过差热分析法(differential thermal analysis,DTA)测试发现,使用ZrO2材质进行球磨时,制备的Li7P3S11具有更高的玻璃化转变温度和晶化温度,表明ZrO2材质球磨有利于形成更为均匀的玻璃态Li7P3S11。实验室条件下,采用机械球磨路径制备玻璃-陶瓷态Li7P3S11电解质,通常采用体积较小的球磨罐(45 mL),并且转数一般高于 500 r·min-1[23,41-42]。Janek 等[43]使用 500 mL的ZrO2球磨罐和2 000个直径为4 mm的ZrO2球磨珠,在转速为210 r·min-1和时间为20 h条件下,成功制备了5 g玻璃态Li7P3S11,经过晶化过程后所得电解质材料的最高室温电导率可以达到8.3×10-3S·cm-1;该研究表明大体积球磨罐和较多数量球,可以有效降低球磨的转速,并且能较大规模制备高性能的Li7P3S11电解质,为实现该电解质的实用化提供技术支撑。
机械球磨制备的Li7P3S11通常为电导率较低的玻璃态,必须进一步热处理形成电导率更高的玻璃-陶瓷态Li7P3S11。玻璃态Li7P3S11通过部分晶化形成玻璃-陶瓷态Li7P3S11,控制材料晶化过程的温度和时间极其重要。早在2006年,Tatsumisago等[44]将机械球磨获得的玻璃态Li7P3S11在不同温度下进行热处理,发现其在250℃开始晶化,并且晶态Li7P3S11在360℃仍能稳定存在;550℃加热时产物开始分解形成Li4P2S6相;电导率测试结果表明,加热产物的电导率在250~360℃范围内随温度升高逐渐增加,在360 ℃时具有最高室温电导率(3.2×10-3S·cm-1),在550 ℃具有最低室温电导率(1.1×10-6S·cm-1)。随后在2011年,Tatsumisago等[42]进一步研究了玻璃态Li7P3S11的晶化转变过程。通过测试差示扫描量热法(differential scanning calorimetry,DSC)曲线观察到216℃处的吸热峰和254℃处的放热峰,分别对应玻璃态Li7P3S11的玻璃化转变温度和结晶温度;该工作分别选取了230、240、250、260 ℃四个温度,研究玻璃化转变过程和结晶过程。通过观察样品的颜色和SEM图像发现,材料在230℃时存在很多空隙并且颜色和室温时相同,表明此时获得的材料为玻璃态;在240℃时空隙消失不见并且材料变得透明,表明开始发生玻璃转变过程;在250℃时空隙和裂缝在材料内部重新出现并且颜色部分透明,表明材料开始部分结晶;在260℃时材料形貌发生较大变化并且完全不透明,表明Li7P3S11晶体已经形成,该研究表明玻璃态Li7P3S11在250~260℃开始结晶化。玻璃态Li7P3S11的热处理时间对最终获得材料的电导率和结构也有很大的影响。Seino等[41]研究了相同温度下,不同热处理时间对玻璃-陶瓷态Li7P3S11的结构和电导率的影响。在300℃下热处理时间分别为1、2、3、240 h时,获得材料的电导率先增加后减小。通过31P MAS-NMR谱图可以明显观察到,热处理时间为240 h时,产生了[PS4]3-和[P2S6]2-对应的化学位移,而处理1、2、3 h的样品仅仅存在[PS4]3-和[P2S7]4-对应的化学位移,表明热处理温度为250~360℃时可以制备玻璃-陶瓷态Li7P3S11,但是过长的热处理时间会使材料分解并伴随着电导率的急剧下降。因此,控制玻璃态Li7P3S11热处理温度和时间是制备高性能玻璃-陶瓷态Li7P3S11的关键因素之一。
2.3 液相合成法
机械球磨法虽然是制备Li7P3S11固态电解质的较为常见的方法,但是该方法具有合成材料的组分不均匀、球磨过程中材料容易粘壁等缺点,因此实现高性能Li7P3S11电解质的大规模制备存在较多的问题。液相法能实现原材料在液态中原子级别的混合,可制备高度均匀的电解质材料,并且该方法容易实现大规模工业化制备和应用于电极材料的表面修饰改性,因此成为制备硫化物固态电解质的研究热点[45]。根据反应过程是否生成沉淀,液相法可以分为2种[46]:(1)溶解-沉淀法,将球磨合成的硫化物电解质完全溶于有机溶剂,如甲醇(MT)、乙醇(EA)、N-甲基甲酰胺(NMF)等,然后采用热处理挥发有机溶剂;(2)悬浮合成方法,将原材料(Li2S、P2S5)加入对其溶解度较低的有机溶剂中形成悬浮颗粒,然后除去有机溶剂,如四氢呋喃(THF)、二甲醚(DME)、乙腈(ACN)等。2种方法后续都需要采用热处理除去挥发性有机溶剂,并对获得产物进行烧结以得到玻璃-陶瓷态Li7P3S11。
目前报道的液相法合成的Li7P3S11多采用的是悬浮合成法,前驱体Li2S、P2S5难以在有机溶剂中完全溶解。2014年,Machida等[47]报道了选取DME作为溶剂,将Li2S、P2S5加入其中搅拌72 h,随后加热除去DME,然后在不同温度下对获得的产物进行热处理。当热处理温度为200、250℃时,通过拉曼谱图分别观察到振动峰位于420 cm-1的[PS4]3-基团和位于405 cm-1的[P2S7]4-基团,同时结合XRD结果确定成功合成了玻璃-陶瓷态Li7P3S11。经过250℃热处理的玻璃-陶瓷态Li7P3S11电解质的室温电导率为2.7×10-4S·cm-1。早期报道的悬浮合成耗时较长,通常为24~76 h。2017年,Tadanaga等[48]在合成过程中引入超声波,大大缩短了合成时间(30 min),成功合成了室温电导率达1.0×10-3S·cm-1、晶粒粒径小于500 nm的玻璃-陶瓷态Li7P3S11。超声波的引入缩短了合成时间,细化了晶粒粒径,并且提高了合成材料的电导率。此外,微波辐射同超声波处理一样也能大幅度缩短合成时间[49]。
3 Li7P3S11电解质的改性研究
3.1 离子电导率改善
相比氧化物和聚合物固态电解质,Li7P3S11的离子电导率具有明显的优势,但与当前的有机电解液的离子电导率相比仍然具有一定的差距。Li7P3S11电解质是通过控制Li2S-P2S5体系的原始物料比制备得到的,而Li2S-P2S5体系根据晶体结构可以分为玻璃态和玻璃-陶瓷态。对于Li2S-P2S5体系的硫化物固态电解质,玻璃-陶瓷态因其结构中生成了介稳的thio-LISICON类似物[50],而具有更高的离子电导率。因此,Li7P3S11电解质的离子电导率改性研究主要是针对玻璃-陶瓷态Li7P3S11电解质。当前,提高Li7P3S11离子电导率的主要策略有通过氧化物[22,51-53]、硫化物[29,54-56]、卤化物[57-59]等掺杂创造缺陷及扩大Li+传输通道、调控玻璃-陶瓷态Li7P3S11结晶度等。此外,通过热压致密化减小材料的晶界[26]、调控玻璃态Li7P3S11的热处理参数[60]等也可以提高离子电导率。
氧化物掺杂(如P2O5[61]、Li2O2[52]、LiNbO3[23]、Nb2O5[23]等)Li7P3S11电解质可以有效提高材料的离子电导率。Hayashi等[61]报道了 P2O5掺杂 70Li·30P2S5改善电解质的离子电导率。他们结合熔融-萃取法和后续热处理合成方法,合成了不同掺杂量的70Li2S·(30-x)P2S5·xP2O5(0≤x≤10)电解质。研究表明,当掺杂量(物质的量分数,下同)为10%时,Li7P3S11电解质具有最高的室温离子电导率(3.0×10-3S·cm-1)和最低的活化能(16 kJ·mol-1)。此外,还采用31P MAS-NMR表征了不同掺杂量下P的化学环境,如图2a所示,分别探测到了位于δ=86的[PS4]3-基团和δ=90处的[P2S7]4-基团;当掺杂量从x=0增加到x=10时,由于O2-取代S2-形成了[POS3]3-基团,其化学位移更接近86,因此观察到峰的部分重叠。该研究结果表明,O2-取代S2-形成了有助于Li+快速传导的[P2OS6]4-基团,从而提高了电解质的离子电导率。为进一步研究O2-的掺杂机理,Shi等[52]采用第一性原理模拟将Li2O2掺杂到Li7P3S11中研究掺杂对离子电导率的影响,得到Li7P3S11-xOx。当掺杂量x=0.75时,模拟发现电导率从未掺杂的5.0×10-2S·cm-1提高到最高值1.09×10-1S·cm-1。该团队选取离子电导率最高的Li7P3S10.25O0.75为研究对象,使用Zeo++软件模拟了掺杂前后O2-对Li7P3S11晶体结构的影响,如图2b所示,发现 O2-取代S2-形成了[P2OS6]4-基团后,距离最近的S2—S3之间的距离从0.363 nm增加到0.396 nm,因此Li+的扩散通道从0.069 48 nm3增加到0.071 38 nm3,进一步证明 O2-掺杂后形成[P2OS6]4-基团,能够拓宽Li+传输通道,进而提升电导率。此外,Tu等[51]通过Nb2O5掺杂来提升Li7P3S11电解质的离子电导率。由于引入具有更大离子半径的Nb5+取代P5+,Li+传输通道被拓宽;同时 Nb5+还会部分取代 Li+,形成Li+空位,两者均会提高离子电导率。
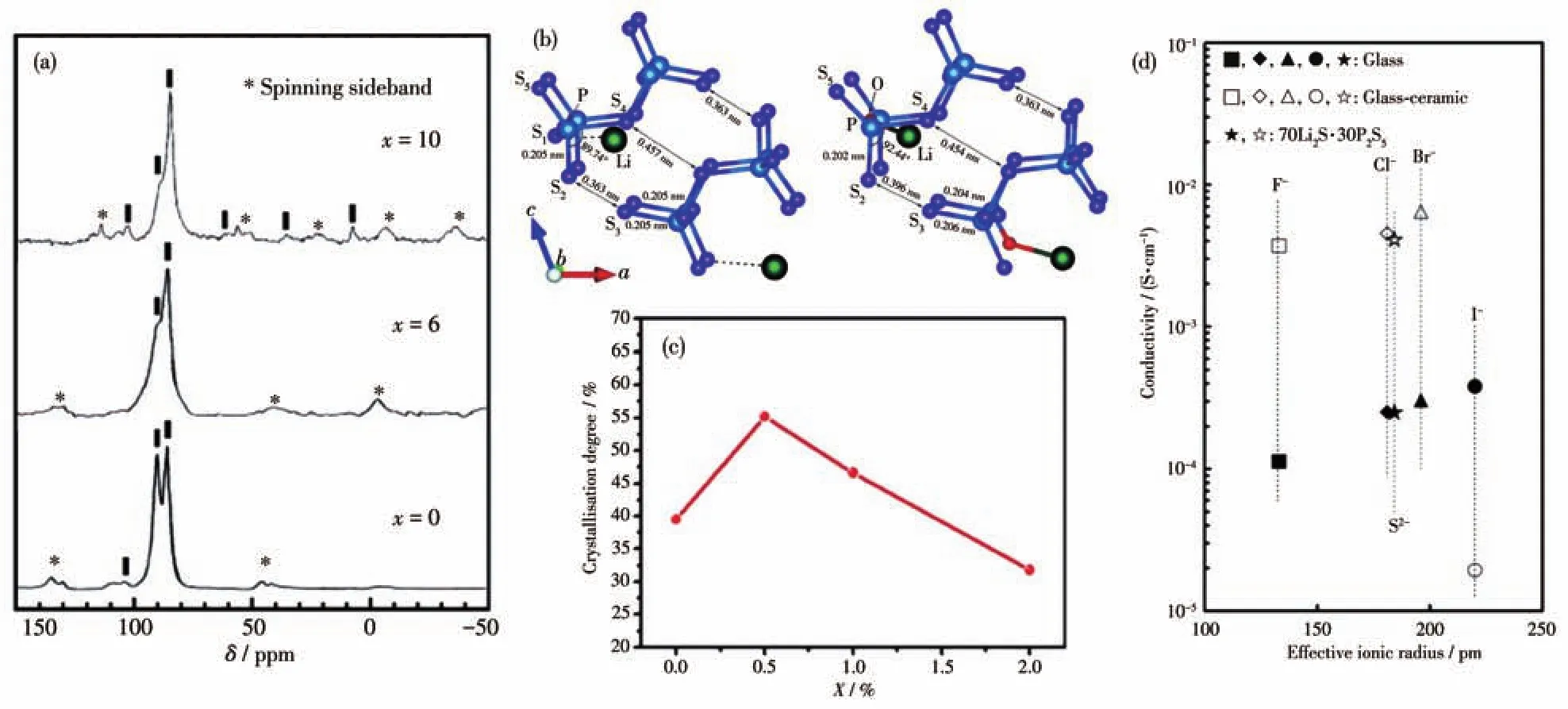
图2 (a)70Li2S·(30-x)P2S5·xP2O5玻璃-陶瓷的31P MAS-NMR谱图[61];(b)Li7P3S11和Li7P3S10.25O0.75中[P2S7]4-双四面体的原子结构[52];(c)玻璃-陶瓷电解质中FeS2掺杂量与结晶度的关系[54];(d)90(0.7Li2S·0.3P2S5)·10LiX的玻璃态、玻璃-陶瓷态在25℃的电导率与卤素的有效离子半径关系及70Li2S-30P2S5玻璃和玻璃-陶瓷电解质的电导率与S2-半径关系[58]Fig.2 (a)31P MAS-NMR spectra of the 70Li2S·(30-x)P2S5·xP2O5glass-ceramics[61];(b)Local atomic structures of[P2S7]4-ditetrahedra in Li7P3S11and Li7P3S10.25O0.75[52];(c)Relationship of the degree of crystallization and the doping amount of FeS2in the glass-ceramic electrolyte[54];(d)Conductivities at 25 ℃ for the 90(0.7Li2S·0.3P2S5)·10LiX glasses and glass-ceramics,shown as a function of effective ionic radius of added halide ion and conductivities changes of the 70Li2S-30P2S5glass and glass-ceramic as a function of the ionic radius of S2-ion[58]
硫化物(如P2S3[40]、GeS2[62]、MnS[63]、WS2[64]、MoS2[56]、FeS2[54]等)也被广泛应用于掺杂改善Li7P3S11电解质的电导率当中。Hayashi等[40]选取P2S3替代部分P2S5来改善Li7P3S11的性能,P3+取代P5+会产生S2-空位得到Li7P3S11-z,当掺杂量从0增加到3%时,离子电导率先增加后减小,在掺杂量为1%时具有最高的离子电导率。为解释掺杂量对Li7P3S11离子电导率的影响,该研究采用31P MAS-NMR表征了不同掺杂量下P—S基团结构,发现掺杂量从0增加到1%时,[PS4]3-基团与[P2S7]4-基团的比例有所增加;当掺杂量增加到2%时,图中出现了[P2S6]4-基团的化学位移,表明生成了电导率较低的Li4P2S6。该研究证明P2S3掺杂能够改变Li7P3S11结构中的[PS4]3-、[P2S7]4-基团的含量。Yang等[54]研究了不同掺杂量的FeS2对Li7P3S11离子电导率的影响。通过对不同掺杂量 (100-x)(70Li2S-30P2S5)-xFeS2(x=0、0.5、1、2)的31P MAS NMR进行分峰,探测到隶属于5种不同P—S基团的核磁信号(玻璃相中的[P2S6]4-对应δ=106,玻璃相中的[P2S7]4-对应δ=90.8,玻璃相中的[PS4]3-对应δ=86,结晶相中[P2S7]4-对应δ=90.8,结晶相中[PS4]3-对应δ=86。通过计算结晶相中[P2S7]4-和[PS4]3-的峰强度之和与5种共振峰强度值之和的比值来粗略估算玻璃-陶瓷态Li7P3S11的结晶度。研究发现,随着掺杂量的变化,Li7P3S11离子电导率先增加后减小,且在掺杂量为0.5%时,具有最高离子电导率2.22×10-3S·cm-1和最高的结晶度55.2%,如图2c所示。
卤化物,如 LiX(X=F、Cl、Br、I)[58]、AgI[57]等,也常用于提高Li7P3S11电解质的离子电导率。Tatsumisago等[58]用不同卤化物 LiX(X=F、Cl、Br、I)掺杂玻璃态Li7P3S11和玻璃-陶瓷态Li7P3S11电解质,如图2d所示。研究发现,对于LiF、LiCl、LiBr掺杂,玻璃态和玻璃-陶瓷态Li7P3S11电解质的离子电导率都随着卤素离子半径增加而变大,而LiI掺杂会导致电解质的离子电导率降低。
掺杂改性是通过掺杂原子改变材料的晶体结构,从而改善Li+在其晶体结构中传导。然而玻璃-陶瓷态Li7P3S11的晶界、空隙[65]也对Li+传输过程产生了极大的影响。Seino等[26]研究了晶界对Li7P3S11电解质的离子电导率影响,如图3a所示,发现冷压后热处理的Li7P3S11的电导率有明显提升(图3b)。SEM结果发现,冷压加热处理后样品的晶界变得难以分辨,说明冷压后热处理能明显减少Li7P3S11的晶界和间隙,从而提升其离子电导率。
3.2 稳定性改善
3.2.1 空气稳定性改善
Li7P3S11电解质对水分很敏感,与空气中水分接触后会迅速产生有毒的H2S气体,导致结构变化和离子电导率衰减,因此其制备过程通常在惰性气氛中进行。提高Li7P3S11电解质的空气稳定性,对降低其生产成本和实现实用化具有重要意义。目前,针对Li2S-P2S5电解质空气稳定性改善的策略主要包括:(1)氧化物掺杂/共混[53,66];(2)基于软硬酸碱理论(HASB),使用较“软”的阳离子(如 Ga3+、Ge4+、Sn4+、Sb4+、Pb4+等)替代 P5+[29];(3)与疏水聚合物复合[67]。
为了改善Li7P3S11电解质的空气稳定性,研究人员做了很多工作。Yang等[23]采用机械球磨法向该电解质结构中掺杂LiNbO3,制备了不同掺杂浓度的Li7-0.06xP3-0.03xNbxS11-0.11xO3x(x=0、0.2、0.4、0.6、1)电解质,并测试了制备的改性电解质暴露在潮湿空气(相对湿度:40%~45%,温度:24~25℃)中H2S气体产量。研究发现,随着O2-掺杂浓度增加,H2S气体逐渐减少,掺杂浓度为x=1的电解质相较于未掺杂的电解质,H2S 气体产速(约为 0.144 cm3·min-1·g-1)减少约89%。该研究表明,O2-掺杂Li7P3S11电解质能够有效改善其空气稳定性。此外,Chen等[67]通过将疏水聚合物-氢化苯乙烯-丁二烯嵌段共聚物(styrene ethylene butylene styrene,SEBS)与 Li7P3S11复合形成Li7P3S11-SEBS复合电解质,测试了不同含量SEBS(质量分数0%、5%、10%)的Li7P3S11-SEBS复合电解质暴露在空气(相对湿度为50%~55%)中的H2S气体产量,如图4a所示,结果表明SEBS含量为5%和10%时,能够大幅度抑制H2S气体产生。此外,如图4b所示,对比了电解质复合前后直接与水接触的变化,发现Li7P3S11电解质遇水后发生分解反应,而Li7P3S11-SEBS复合电解质则能在水中稳定存在,进一步证明了Li7P3S11与疏水聚合物SEBD复合后能明显改善其空气稳定性。

图4 (a)100 mg的Li7P3S11和100 mg的Li7P3S11与疏水SEBS复合样品暴露在一定体积空气中H2S产量随时间变化图[67];(b)Li7P3S11样品浸泡在水中前后变化(完全水解并消失在水中),Li7P3S11与疏水SEBS复合样品侵泡在水中前后变化(依旧保持为膜)[67];(c)Li/Li7P3S11/不锈钢电池和(d)Li/Li6.988P2.994Nb0.2S10.934O0.6/不锈钢电池的循环伏安图(电压范围:-0.5~0.5 V(vs Li/Li+)、扫速:1 mV·s-1)[23]Fig.4 (a)H2S amount released as a function of time for fixed volume air exposed to 100 mg of bare Li7P3S11and 100 mg of composite with hydrophobic SEBS polymer[67];(b)Bare Li7P3S11before and after flooding in water,showing full hydrolysis and disappearance in water,composite electrolyte film before and after flooding in water,showing retention of the film[67];Cyclic voltammetry curves of(c)Li/Li7P3S11/stainless steel and(d)Li/Li6.988P2.994Nb0.2S10.934O0.6/stainless steel cells with the voltage range from-0.5 to 5 V(vs Li/Li+)at 1 mV·s-1[23]
3.2.2 电化学稳定性改善
固态电解质与Li金属负极、高电压正极材料结合组装固态电池能有效地提高电池的能量密度,这就要求固态电解质在充/放电过程中具有足够高的电化学稳定性,即具有较宽的电化学窗口。固态电解质在超过其电化学窗口运行时,会使得电解质/电极界面发生副反应,并且副反应产物会阻碍Li+传输[68],因此准确测试固态电解质的电化学窗口也很重要。
硫化物固态电解质的电化学窗口通常低于氧化物固态电解质,主要由于O2-相较于S2-更难被氧化。Mo等[69]通过第一性原理计算,发现硫化物固态电解质的本征电化学窗口为1.7~2.5 V(vs Li/Li+),其中Li7P3S11电解质的电化学窗口在2.28~2.31 V(vs Li/Li+)。然而实际测试的电化学窗口远大于理论计算值,甚至高达0~10 V(vs Li/Li+)[59],造成这种现象的主要原因是传统测试方法的限制。Han等[70]研究发现,使用Li/固态电解质/阻塞电极(Pt或不锈钢)装置进行循环伏安法测试时,电极与电解质的接触面积过小会限制反应动力学,同时电解质/电极界面也会形成中间层阻碍Li+传输。因此,实验所测得的固态电解质电化学窗口通常远低于其本征值。
准确测试电解质的电化学窗口固然重要,但是提升电解质在运行电压窗口内的稳定性更加重要。目前用循环伏安法测试Li7P3S11电解质的电化学稳定性,电压范围通常设定为-0.5~5 V(vs Li/Li+)[29,54,57,63],这表明Li7P3S11电解质对锂金属的稳定电压可高达5 V(vs Li/Li+)。虽然Li7P3S11电解质的电化学稳定性测试窗口多在-0.5~5 V(vs Li/Li+),但在其循环伏安图中可以观察到除Li金属的阴极电流和Li+的阳极电流之外,还探测到Li7P3S11电解质微弱的氧化/还原电流,这表明 Li7P3S11在-0.5~5 V(vs Li/Li+)内并不能稳定存在。Yang等[23]向Li7P3S11电解质中掺入LiNbO3,如图4c、4d所示,发现未掺杂的Li7P3S11的循环伏安图中可以观察到Li金属的阴极电流、Li+的阳极电流以及对应于2.1 V(vs Li/Li+)处小的阴极电流,表明Li7P3S11电解质在该电压窗口范围内不稳定,发生了氧化反应。而O2-掺杂的电解质在相同测试条件下,几乎观察不到除了Li金属的阴极电流和Li+的阳极电流之外的任何其他电流,表明O2-掺杂能明显改善Li7P3S11电解质的电化学稳定性。此外,卤化物掺杂也被证明能明显提升Li7P3S11的电化学稳定性。Ryu等[57]通过机械球磨法制备了AgI掺杂的Li7P3S11电解质,通过对比掺杂前后的循环伏安图,发现掺杂后的Li7Ag0.1P3S11I0.1电解质的电化学稳定性也能明显改善。
4 基于Li7P3S11电解质的全固态电池
由于Li7P3S11电解质的高室温离子电导率,该电解质被广泛应用于构筑高性能的全固态电池。根据正极活性物质种类可将Li7P3S11基全固态电池分为全固态锂硫电池和全固态锂电池。Li7P3S11电解质具有的较高反应活性使得构筑高性能全固态电池面临较大挑战,主要包括与正极相关的固固界面空间电荷层效应[71]、元素相互扩散[72]、活性物质与电解质固固接触失效[73]和与负极相关的锂枝晶生长、锂金属诱导固态电解质还原、锂金属体积变化[74]等。因此,构筑结构稳定且有利于Li+传导的固态电解质/电极界面也成为当前固态电池的研究重点。基于Li7P3S11电解质构筑全固态电池具有重要意义,图5给出了全固态电池从材料合成、复合电极制备到电芯设计的流程图。

图5 基于Li7P3S11电解质的全固态电池构筑示意图Fig.5 Schematic diagram of all-solid-state battery based on Li7P3S11electrolyte
4.1 高性能全固态锂硫电池构筑
全固态锂硫电池的正极材料通常由硫单质、过渡金属硫化物、硫化锂等活性物质、电解质和导电剂按一定比例混合合成,负极通常采用Li、Li-In合金等。基于固态电解质的全固态锂硫电池能有效抑制传统液态锂硫电池中多硫化锂的穿梭效应。在固态锂硫电池中,电池的性能通常受材料的结晶度、正极组分的颗粒大小以及固固接触等影响,通常采用机械球磨混合正极材料实现各组成的紧密接触,从而构筑高效的电子/离子导电网络。
4.1.1 单质硫基全固态锂硫电池
硫因具有极高的理论比容量(1 672 mAh·g-1)和能量密度(2 500 Wh·kg-1)[75],有望成为下一代储能电池的正极材料。传统液态锂硫电池面临很多挑战,如穿梭效应、低电子电导率、剧烈的体积膨胀等问题,而固态电解质可以避免多硫化物溶解,从而解决穿梭效应;低的电子电导率可通过在正极加入碳导电剂来改善。Tu等[56]通过混合硫单质、乙炔碳黑、Li7P2.9S10.85Mo0.01电解质(质量比 3∶1∶6)制备了S-Li7P2.9S10.85Mo0.01-C复合正极材料,结合金属锂负极材料和Li7P2.9S10.85Mo0.01电解质,在0.05C倍率下实现了较高的首圈放电比容量(1 020 mAh·g-1),并且30圈循环后具有较高的容量保持率。硫单质在充放电过程中的体积变化问题,会导致固态电池的循环、倍率性能变差,巧妙设计正极结构是常用的解决体积变化问题的手段之一。Zhang等[75]使用具有多孔结构的BP2000碳材料和S单质,制备出的S@BP2000具有核-壳结构(图6a);如图6b所示,其中S@BP2000与Li7P3S11电解质质量比为2∶3的复合正极材料在3C倍率下,首圈放电比容量高达1 356 mAh·g-1(硫理论容量的81%),循环1 200圈后仍能保持985.3 mAh·g-1的放电容量和较高的库仑效率。全固态锂硫电池中正极部分固固接触、活性物质的颗粒尺寸等对电池的循环性能也有重要影响。Akbulut等[76]用3种不同合成方法制备还原氧化石墨烯/硫单质复合材料,如图6c所示,将其制备成氧化石墨烯、导电碳、固态电解质、硫单质质量比为1∶1∶1∶1的复合正极。第1种方法是通过熔融扩散法将氧化石墨烯/硫单质复合,另外2种方法都是基于液相法,区别在于硫源不同,二者分别采用硫单质和还原Na2S2O3获得的硫单质作为硫源。通过SEM观察到液相法制备的氧化石墨烯/硫单质复合材料中的硫单质具有更小的粒径并且与氧化石墨烯接触更为紧密。第3种方法制备的复合正极材料中硫单质粒径最小,且组装的固态锂硫电池循环性能最好(图6d)。该研究表明活性物质的粒径大小、复合正极中固固接触好坏对于以硫元素作为活性物质的全固态锂硫电池性能具有重要影响。

图6 (a)S@BP2000纳米复合材料的TEM图[75];(b)S@SBP2000复合正极构筑的全固态电池在室温下3C倍率循环图,容量计算基于S的质量[75];(c)三种正极材料的合成路线图:熔融扩散法(正极-1号),溶液法(正极-2号、正极-3号)[76];(d)不同正极材料循环过程中的电池容量衰减图[76]Fig.6 (a)TEM images of S@BP2000 nanocomposite[75];(b)Cycle performance at 3C rate capabilities of S@SBP2000 cathode for all-solid-state battery at RT(Capacities were calculated based on the weight of sulfur)[75];(c)Schematic synthesis routes of three cathodes:melt diffusion technique(cathode no.1)and solution-based techniques(cathode no.2 and cathode no.3 respectively)[76];(d)Capacity decays of cells with different cathodes upon cycling[76]
4.1.2 硫化锂基全固态锂硫电池
硫化锂较高的理论比容量(1 170 mAh·g-1)也使得其成为一种潜在的全固态锂硫电池正极材料。Yu等[65]将商业化的Li2S在较高转速下球磨获得纳米级Li2S,将其与Li7P3S11电解质、导电碳黑、碳纳米纤维按照4∶4∶1.5∶0.5的质量比混合制备复合正极,Li-In合金作为负极,组装Li2S/Li7P3S11/Li-In全固态锂硫电池,在0.064 mA·cm-2的电流密度下,首圈放电比容量高达1 139 mAh·g-1,并且循环30圈后仍维持850.0 mAh·g-1的放电比容量。
4.1.3 过渡金属硫化物基全固态锂硫电池
过渡金属硫化物与硫化物固态电解质具有相近的电化学势,因此它们之间兼容性较好,可以避免空间电荷层的形成[77],是常用的一种全固态锂硫电池正极材料。减小正极活性物质的尺寸通常是提高这类电极材料电化学性能的有效方法之一,然而纳米级活性物质与微米尺寸硫化物固态电解质的固固接触仍然是一大挑战。Xu等[78]通过液相法在硫化钴纳米片上原位生长Li7P3S11电解质材料,如图7a所示,通过TEM发现原位生长的Li7P3S11颗粒尺寸仅为10 nm,这种结构设计能实现固态电解质与活性物质的紧密接触和硫化钴活性物质循环过程中均匀的体积变化。长循环测试结果表明,如图7b所示,在0.38 mA·cm-2的电流密度下,硫化钴/Li7P3S11复合正极在循环50圈后仍有91.9%的容量保持率,而硫化钴纳米片正极在相同条件下容量保持率仅有21.1%,表明这种结构设计能够明显改善电池的循环性能。虽然过渡金属硫化物与硫化物电解质具有较好的兼容性,但是两者间的界面阻抗仍较大,因此减小界面阻抗也是构筑高性能过渡金属硫化物基全固态电池的一大挑战。Tu等[79]通过液相法合成了Li7P3S11包覆MoS2的复合正极材料,显著减小了活性物质和电解质之间的界面阻抗,提高了全固态锂硫电池的性能。

图7 (a)硫化钴、硫化钴-Li7P3S11纳米复合材料、Li7P3S11电解质的合成策略示意图;(b)硫化钴-Li7P3S11纳米复合材料和硫化钴纳米片电极在电流密度为0.38 mA·cm-2下的放电-充电循环性能图[78]Fig.7 (a)Schematic illustration of the synthesis strategy for cobalt sulfide,cobalt sulfide-Li7P3S11nanocomposites,and neat Li7P3S11electrolyte;(b)Cycle performances of the cobalt sulfide-Li7P3S11nanocomposite and cobalt sulfide nanosheet electrodes discharged and charged at a constant current density of 0.38 mA·cm-2[78]
4.2 高性能全固态锂电池构筑
常用的商业化全固态锂电正极材料一般为含锂过渡金属氧化物,如LiMnxNiyCozO2(x+y+z=1)、LiCoO2、LiMn2O4等。由于含锂氧化物与硫化物之间存在较大的化学势差,会驱动Li+从硫化物固态电解质移向氧化物正极直至平衡,形成空间电荷层。空间电荷层的形成会使得Li+在氧化物正极/硫化物电解质的界面传导变得困难,导致界面阻抗增加[80]。氧化物正极与硫化物固态电解质之间还会发生元素扩散[72],在界面处形成离子电导率较低的中间相,阻碍Li+在界面处的传导。在传统液态锂离子电池中,由于有机电解液良好的浸润性使得活性材料与电解液具有较好的接触,锂离子能在两者间以较小的阻碍传导。然而,在全固态电池中,正极材料与固态电解质间为固固点接触,导致Li+在两者间传导变得更加困难;此外,充放电过程中正极材料会发生体积变化,导致部分活性物质/电解质的接触界面失效,正极部分有效接触减少,从而影响正极内部的锂离子传导。上述全固态锂电池中诸多的固固界面问题,会导致成分间界面阻抗增加、电池容量损失、循环性能变差等问题。
LiCoO2具有较高的理论比容量(274 mAh·g-1)和良好的电子导电性,常用于全固态锂电池正极材料。Xu等[64]研究了不同比例WS2和LiBr共掺杂的Li7P3S11电解质与LiCoO2正极材料的稳定性,如图8a所示,由Heinrich Lenz-capacitor-resistance(LCR)数字电桥的介电测试图看出WS2和LiBr共掺杂后的Li7P3S11与LiCoO2的界面电容明显减小,表明电解质共掺杂改性可以显著减小Li7P3S11电解质和LiCoO2正极材料的空间电荷效应,并且其组装而成的全固态电池的循环性能有明显改善(图8c)。目前克服空间电荷效应的主要策略是对含锂过渡金属氧化物进行包覆改性,如LiNbO3[10]、Al2O3[81]、LiTaO3[82]等对LiCoO2正极进行包覆。这些包覆材料不仅具有宽的电化学窗口,而且与硫化物电解质具有较好的兼容性。Wang等[10]使用LiNbO3包覆的LiCoO2正极材料结合Li7P3S11电解质和LiF包覆的金属锂负极构建全固态锂电池,如图8b所示,在0.1 mA·cm-2的充放电电流密度下,首圈放电比容量为118.9 mAh·g-1,100圈循环后仍具有81.4%的容量保持率。
富镍层状正极材料Li[Ni1-xMx]O2(M=Co、Mn、Al等)因具有高的理论比容量(200 mAh·g-1)和工作电压平台,而成为构筑高能量密度的全固态锂电池的理想候选正极材料。镍钴铝酸锂Li[Ni0.8Co0.15Al0.05]O2(NCA)正极材料是通过Co和Al部分取代Ni而得到,这种取代提高了正极材料的结构稳定性和电池的安全性能[83]。Park等[28]报道了LiMoO3-LiI包覆的NCA正极材料在Li7P3S11基全固态锂电池中的应用,如图8d所示,发现LiMoO3-LiI包覆后能显著提升全固态电池的容量,这是因为包覆层能够有效抑制活性材料与固态电解质的副反应。镍钴锰酸锂(如LiNi0.6Co0.2Mn0.2O2、LiNi1/3Co1/3Mn1/3O2、LiNi0.8Co0.1Mn0.1O2等)也常被选取为正极材料应用于全固态锂电池中。Ryu 等[57]将 LiNbO3包覆的 LiNi0.6Co0.2Mn0.2O2、Li7P3S11、导电碳黑按照70∶28∶2的质量比混合,制备出复合正极,并将其与Li7Ag0.1P3S11I0.1固态电解质和金属In负极结合构筑全固态锂电池。如图8e所示,该电池在1C充放电倍率下,首圈放电比容量为76.25 mAh·g-1,循环 10 圈后仍具有 45.74 mAh·g-1的放电比容量。

图8 (a)四个样品的电容与频率关系[64];(b)全固态电池在电流密度为0.1 mA·cm-2的循环性能[10];(c)不同玻璃-陶瓷电解质组装固态电池循环性能[64];(d)基于Li7P3S11电解质的全固态电池充放电曲线[28];(e)全固态电池In/Li7Ag0.1P3S11I0.1/LiNi0.6Co0.2Mn0.2O2在1C电流密度下的充放电曲线[57]Fig.8 (a)Dependence of the series capacitances of four samples on frequency[64];(b)Cycling performance of assembled all-solid-state battery at 0.1 mA·cm-2[10];(c)Cycling performances of the all-solid-state battery assembled with different glass-ceramics[64];(d)Charge-discharge profiles of all-solid-state battery using Li7P3S11electrolytes[28];(e)Charge-discharge curves of the In/Li7Ag0.1P3S11I0.1/LiNi0.6Co0.2Mn0.2O2cell at the current rate of 1C[57]
5 总结和展望
全固态电池因其高的本征安全性和高能量密度,受到越来越多研究人员的关注。Li7P3S11具有接近液态电解质的电导率,并且合成原材料相对其他硫化物电解质价格低廉,成为一种具有较好应用前景的固态电解质。但是其商业化应用仍面临很多挑战。本文综述了Li7P3S11的晶体结构与锂离子传导机理,有助于研究人员进一步改善电解质的综合性能;同时还总结了现有的合成路径、电导率提升策略、稳定性改善方法以及高性能全固态电池构筑策略等,为后续研究人员提供关于Li7P3S11电解质的基本研究框架。我们认为后续关于Li7P3S11电解质的研究还有很多难题需要克服:
(1)空气稳定性优化。Li7P3S11电解质在空气中不稳定,极易与水发生反应,导致制备过程通常要在惰性范围内进行,造成电解质制备成本上升;此外,还导致后续全固态电池制造过程工艺繁琐,对环境的要求提升,进一步提高了全固态电池的生产成本,阻碍了Li7P3S11基全固态电池的实用化进程。
(2)正极/电解质界面修饰。Li7P3S11作为硫化物固态电解质的一种,也面临其他硫化物全固态电池中的挑战,如空间电荷层效应、元素互扩散、活性物质与电解质固固接触失效等。针对上述挑战,当前正极/电解质界面修饰手段主要包括:(ⅰ)氧化物包覆正极材料,即在氧化物活性物质表面包覆LiNbO3、Li4Ti5O12、LiTaO3、Al2O3等。这些氧化物具有与硫化物兼容性较好、电化学窗口宽等特点,因此,在一定程度上可以减缓空间电荷层效应和元素互扩散。(ⅱ)硫化物电解质包覆正极材料。通过液相法包覆硫化物电解质可以提升电解质和活性物质的接触面积,缩短Li+的传输距离,在一定程度上还可以缓解正极材料体积效应所带来的固固接触失效。(ⅲ)电解质改性。将硫化物电解质中部分硫原子以氧原子取代,改善硫化物与氧化物正极材料的兼容性,提升正极/电解质界面稳定性。
(3)负极界面修饰。锂金属的高理论比容量(3 860 mAh·g-1)可以进一步提高全固态电池的能量密度,然而硫化物电解质与锂金属的副反应和锂枝晶的生长,使得硫化物全固态锂金属电池的应用较难。目前负极/界面改性主要集中在:(ⅰ)原位生成固体电解质界面膜。近年来,很多工作报道了在锂金属负极表面原位生成LiI、LiF等界面层,在一定程度上可以抑制锂枝晶的生长;同时,界面层的电子绝缘特性也会抑制硫化物与锂金属之间的副反应。(ⅱ)电解质组分调控。对硫化物电解质掺杂改性或者物理共混可以提升硫化物电解质与锂金属负极界面稳定性。(ⅲ)锂金属合金化。有大量文献报道Li-In、Li-Sn、Li-Al合金作为负极可以抑制锂枝晶的生长,因此锂金属合金化也是一种有效的负极界面修饰手段。
(4)全气候条件下应用。全固态电池的高低温性能已经有很多学者报道,然而Li7P3S11基全固态电池的高低温性能研究比较匮乏。研究其高低温性能及其背后的工作机理,探索极端气候条件下的应用,也将大大推进Li7P3S11电解质的实用化。
猜你喜欢
卫星电视与宽带多媒体(2022年18期)2022-10-26
干旱地区农业研究(2022年3期)2022-05-24
矿冶工程(2021年4期)2021-09-15
消费导刊(2019年5期)2019-08-22
山西果树(2017年4期)2018-02-08
科技创新导报(2017年2期)2017-04-12
中国新技术新产品(2017年2期)2017-01-20
绿色科技(2016年20期)2016-12-27
科教导刊·电子版(2016年3期)2016-03-14
湖北农业科学(2014年13期)2014-08-28
