
疏水性含钒多酸高效催化氧化脱硫
2022-06-09 07:38宋宇飞连丽飞张利民陈伟
山西大学学报(自然科学版) 2022年3期
宋宇飞,连丽飞,张利民,陈伟
(北京化工大学 化工资源有效利用国家重点实验室,北京 100029)
0 引言
原油和石油馏分中的硫元素通常以单质硫、硫化氢和多种有机硫(硫醇、硫醚、环硫醚、二硫化物、噻吩及其同系物)的形式存在[1]。硫的存在不仅严重危害原油加工过程,造成设备腐蚀。同时,在成品油燃烧使用过程中,含硫物质燃烧会产生大量SOx气体,不仅会引起酸雨,还会在大气中形成微小颗粒物,严重危害自然环境和人类健康。由于含硫物质的极大危害性,世界各国对成品油中的含硫量设定了极为严格的标准[2]。早在2006年,美国环保部规定柴油中含硫量应低于15 μg/g,欧盟的柴油标准中要求硫含量低于10 μg/g。我国从2003年开始,不断提高柴油产品质量,严格控制硫含量。2017年供应的普通柴油(国五车用柴油)中要求硫含量应低于10 μg/g,2021年7月我国重型柴油车已经开始执行更为严格的国六排放标准。不断降低成品油中的硫含量,提升油品质量,能有效降低污染排放,保护生态环境,提高城市居民空气质量,这使得深度脱硫成为一个全球性的紧迫的任务[3-4]。
柴油中总硫含量的80%是噻吩硫化物,这其中主要是噻吩(BT)、二苯并噻吩(DBT)以及 4,6-二甲基二苯并噻吩(4,6-DMDBT)[5]。目前工业上多采取的催化加氢脱硫技术和非加氢脱硫技术[6]。催化加氢脱硫能高效地去除硫醇等含硫物质,但对DBT及其衍生物等芳香性硫化合物的脱除效果较差,同时,苛刻的运转条件(反应温度300℃~400℃、氢气的使用以及20 atm~100 atm的氢气反应压力)造成脱硫成本高以及脱硫后辛烷值损失较大,极大限制了其应用。而氧化脱硫技术由于反应温度低,操作过程绿色环保,受到了越来越多的关注。氧化脱硫和萃取脱硫耦合技术被认为是最有前景的催化脱硫体系,它的主要过程是油品中有机硫化物被选择性地氧化为其相应的亚砜和砜,然后产生的亚砜和砜在后续的萃取过程中被直接去除[7-8]。离子液体[9-11],作为“绿色”溶剂,有较好的稳定性、非爆炸性、可回收再利用、易于处理等特点。离子液体能从汽油和柴油中有效萃取单环芳烃和多环芳烃的硫和氮化合物,相比于传统的溶剂展现出更多优越的性能,被广泛应用到油品脱硫的研究中。
多金属氧酸盐(POM)是一类前过渡金属(钒、钼、钨、铌等)的金属氧簇化合物,它们表现出强的 Brønsted 酸性[12-15]。近年来,杂多酸(盐)作为有机合成和石油化工中的催化剂已经愈来愈受到人们的关注。它具有确定的结构,既有配合物和金属氧化物的结构特征,又有酸性和氧化还原性能,既可作为均相或多相催化剂,又可作为同时传递质子和电子的双功能催化剂。本课题组从2008年开始开展多酸脱硫的基础研究工作。2012年,首次采用Na9EuW10O36·32H2O 和 Na9LaW10O36·32H2O 为 催化剂、H2O2作为氧化剂、1-丁基-3-甲基咪唑四氟硼酸盐为萃取剂,催化氧化脱除模拟油中的含 硫 底 物 DBT、BT 和 4,6-DMDBT[16]。 在30℃下,当催化体系中H2O2/DBT/LaW10的摩尔比为 500∶100∶5时,25 min就达到 99.9% 的深度脱硫。随后,利用功能表面活性剂分子调控杂多酸结构,构筑了三相微乳体系,微乳体系可以有效促进含硫底物的萃取过程,大大加速了反应进程[17]。为了进一步解决杂多酸流失等问题,课题组先后采用介孔二氧化硅[18]、Tris-LDH[19]、γ-氧化铝[20]等多种载体,深入探索了静电相互作用、主客体层板相互作用以及共价键相互作用等不同多酸负载型催化剂的氧化脱硫性能。经过十余年的持续研究,围绕氧化脱硫体系取得了一定的工作积累。
在前期工作基础之上,本文提出构筑疏水性含钒多酸催化剂(图1),探索其催化氧化脱硫性能。主要基于以下两个方面考虑:1)含钒多酸催化剂在氧化催化反应中具有高效的催化性能[21],可氧化含硫底物,采用含钒杂多酸替代稀土杂多酸,可有效降低催化剂成本;2)采用不同链长的有机胺阳离子,构筑新型疏水性含钒多酸催化剂,有机疏水长链可有效调控催化剂的疏水性能,促进含硫底物与催化剂活性中心的可接近性。因此,本文构筑了六种不同链长的疏水性含钒多酸催化材料,详细探究了其萃取催化氧化脱硫性能。实验结果显示:(DDA)6PW9V3催化剂在30℃条件下,25 min内即可实现99.9%以上的脱硫效率,当温度增加到50℃时,仅需10 min就可以实现99.9%深度脱硫效果。
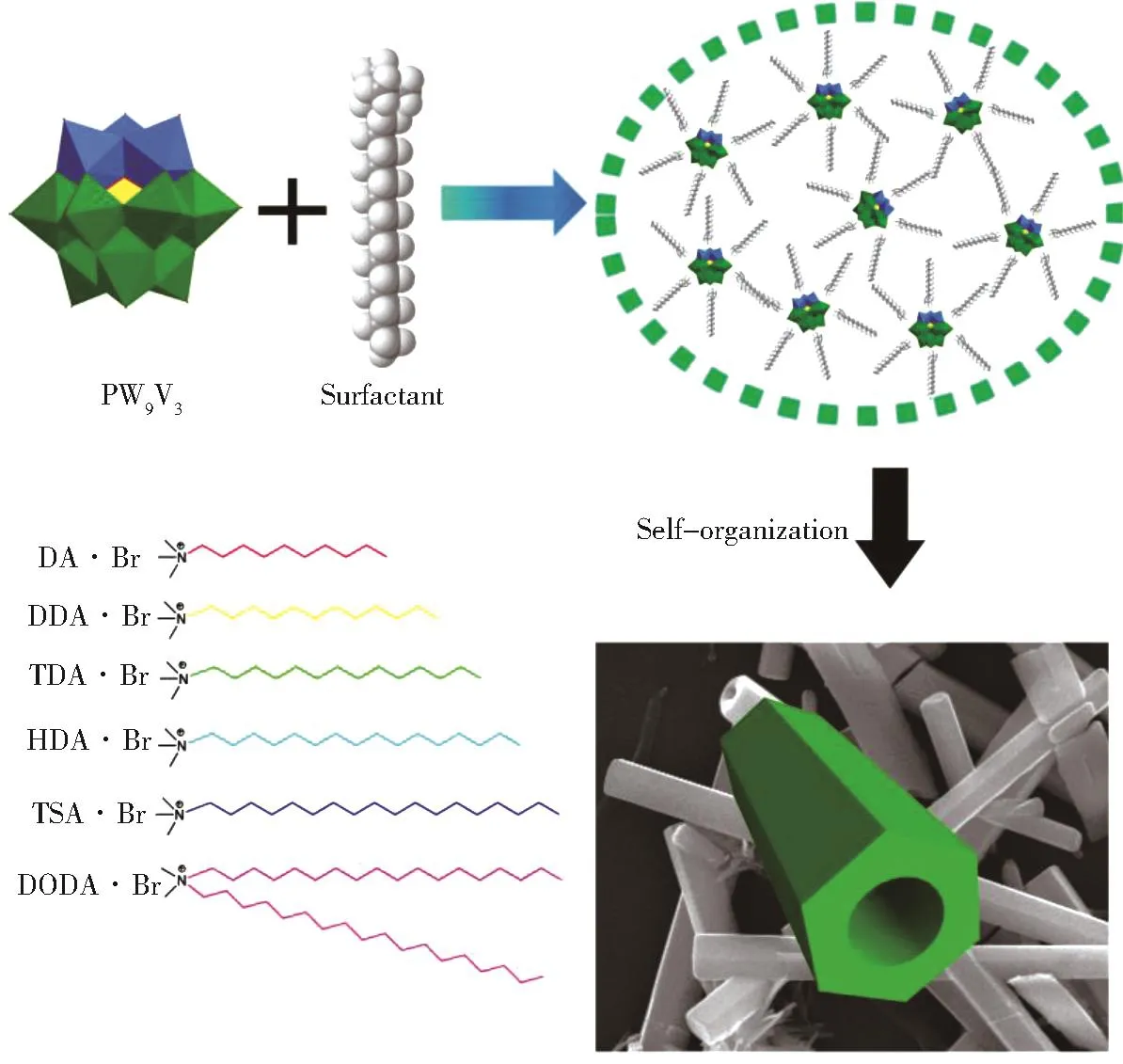
图1 离子交换策略制备疏水性含钒多酸流程示意图Fig.1 Schematic diagram of the preparation of hydrophobic vanadium-containing polyoxometalate by ion-exchange strategy
1 实验过程
1.1 药品与测试条件
K6[PW9V3O40]•4H2O[22]、Cs5[PW10V2O40][22]、H6[PMo9V3O40][23]、H5[PMo10V2O40][23]和 H4[PMo11VO40][24]参 照 文 献 合 成 。 二 苯 并 噻 吩(DBT,98%)、苯并噻吩(BT,98%)、4,6-二甲基二苯并噻吩(4,6-DMDBT,98%)、双氧水(H2O2,质量分数30%)、正辛烷(99%)、十烷基三甲基溴化铵(DA•Br)、十二烷基三甲基溴化铵(DDA•Br)、十四烷基三甲基溴化铵(TDA•Br)、十六烷基三甲基溴化铵(HDA•Br)、十八烷基三甲基溴化铵(TSA•Br)和二甲基双二十八烷基溴化铵(DODA•Br)等化学药品从Alfa Aesa公司购买。离子液体1-甲基-3-辛基咪唑六氟磷酸盐([omim]PF6)从百灵威公司购买。乙腈、氯仿和乙醇等溶剂从北京化工试剂厂购买。所有试剂购买后未做任何纯化处理,直接使用。
1.2 催化材料制备方法
(DDA)6PW9V3催化剂的合成过程
称取1倍当量的K6PW9V3O40•4H2O溶解于去离子水中,再称取6倍当量的DDA•Br溶解在氯仿溶剂中,将两种溶剂混合。混合后,氯仿体系立即出现浑浊,用机械搅拌器剧烈搅拌3 h。反应结束后,静置 30 min,用分液漏斗分出下层氯仿溶液,旋干溶剂后,将得到的产品在乙腈与水的混合溶液(乙腈∶水体积比3∶1)中溶解,静置一段时间,得到具有固定形貌的催化剂,在50℃条件下真空干燥5 h,得到最终产品。(DA)6PW9V3、(TDA)6PW9V3、(HAD)6PW9V3、(TSA)6PW9V3和(DODA)6PW9V3的合成过程与(DDA)6PW9V3过程一致,只是将反应原料中DDA•Br替换成相应的溴化盐。
1.3 催化氧化脱硫实验
模拟油的配制:配制含硫量为1 000 μg/g的模拟油,具体操作过程如下:称取2.93 g的DBT溶解在200 mL的正辛烷溶液中,随后转移到500 mL的容量瓶内定容。1 000 μg/g BT模拟油和1 000 μg/g 4,6-DMDBT模拟油采用同样的操作方法[18]。
将规格为50 mL的单口烧瓶放到已经控制好反应温度(30℃、40℃或50℃)的油浴中,随后,依次向烧瓶中加入特定质量的催化剂、1 mL离子液体、10 mL模拟油。搅拌10 s后,开始加入特定含量的双氧水,开始计时。每隔5 min取一次样,每次静置后取出上层清液用气相色谱分析[18]。
2 实验结果
2.1 催化剂结构表征
为了验证含钒多酸与表面活性剂分子是否交换成功,以及含钒多酸阴离子的结构稳定性,我们采用 FT-IR、1H NMR、31P NMR、热重以及元素含量分析等表征手段认识不同化合物(DDA)6PW9V3、(DA)6PW9V3、(TDA)6PW9V3、(HDA)6PW9V3、(TSA)6PW9V3和(DODA)6PW9V3的 结 构(图S1-S3)。为了避免赘述,我们以化合物(DDA)6PW9V3为例进行细致讨论。
图 2(a)展示的是化合物 DDA·Br、PW9V3和(DDA)6PW9V3红外光谱图,从DDA·Br红外图中,可以看到在2 848 cm-1和2 917 cm-1处出现较强的吸收峰,分别属于−CH2−的对称和不对称吸收峰;PW9V3的红外谱图在1 070 cm-1、960 cm-1、878 cm-1以及792 cm-1处出现较强的吸收峰,分别可归属为[PW9V3O40]6-阴离子中P−Od键的伸缩振动、V=Oa键的伸缩振动、W−Ob−W键的不对称振动以及V−Oc−V键的不对称振动[22]。将DDA·Br与PW9V3离子交换后,得到的化合物(DDA)6PW9V3的红外光谱图中均出现典型的−CH2−和[PW9V3O40]6-阴离子化学键的振动信号峰。其中−CH2−的信号峰由原先的2 917 cm-1和2 848 cm-1偏移到2 919 cm-1和 2 850 cm-1,[PW9V3O40]6-阴离子的信号峰则 分 别 偏 移 到 1 054 cm-1、956 cm-1、878 cm-1、793 cm-1等位置。其他化合物的红外谱图与(DDA)6PW9V3类似,均发生了较为明显的位移变 化(见 图 S1)。 对 比 DDA·Br和(DDA)6PW9V3的核磁氢谱图(图 2(b)、图 S2)可以看出,离子交换前后,核磁氢谱中在0.86(−CH2−CH3)、1.26(−CH2−)、1.67(−CH2−CH3)出现的三组峰的化学位移没有发生明显的变化,N+−CH2−和N+−CH3的化学位移则由交换前的3.04和3.26分别移动到了3.26和3.37。这表明离子交换后,N+位置的化学环境发生了明显的变化,这说明DDA+有机链与[PW9V3O40]6-阴离子之间具有较强的静电相互作用。与此同时,交换后,(DDA)6PW9V3的核磁31P谱在−13.20处出现一个峰(图2(c)),表明[PW9V3O40]6-阴离子在与 DDA+有机链结合后,仍然保持较为完整的结构[22]。
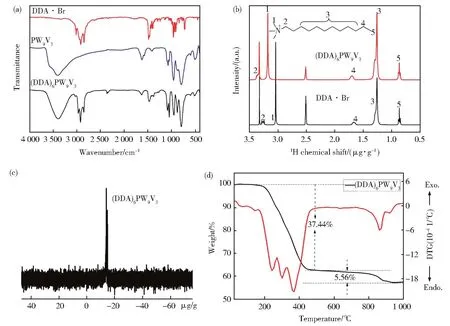
图2 (a)化合物DDA·Br、PW9V3和(DDA)6PW9V3红外光谱图;(b)化合物DDA·Br和(DDA)6PW9V3核磁氢谱图(d6-DMSO);(c)化合物(DDA)6PW9V3的核磁31P谱图(d6-DMSO);(d)化合物(DDA)6PW9V3热重分析图谱Fig.2 (a)FT-IR spectra of DDA·Br,PW9V3,and(DDA)6PW9V3;b)1H NMR spectra of DDA•Br and(DDA)6PW9V3in d6-DMSO;c)31P-NMR spectra of(DDA)6PW9V3in d6-DMSO;d)TGAof(DDA)6PW9V3
(DDA)6PW9V3的热重图谱(图2(d))显示两个较为明显的失重,第一个失重出现在212℃到515℃之间,质量降低了37.44%,归因于分子内有机组分DDA+的热降解;第二个失重从694℃到920℃,质量进一步减少了5.56%,归因于多酸团簇的部分热降解。热重结果也进一步表明化合物(DDA)6PW9V3在200℃以下具有良好的热稳定性,能够在后续开展的萃取催化氧化脱硫反应中保持结构稳定。其他化合物的热损失与(DDA)6PW9V3相似(图S3)。通过有机元素的分析测试(表S1)显示了催化剂的组成元素C、H和N的测试值与理论值基本相同。并且通过(DDA)6PW9V3的C、H、N的元素分析可以得出分子内有机组分DDA+的质量百分比是35.63%,这一结果与(DDA)6PW9V3的第一阶段热损失37.44%相吻合。
扫描电子显微镜(SEM)和透射电子显微镜(TEM)的结果显示化合物(DDA)6PW9V3在乙腈和水的混合物体系中呈现规则的中空六棱管形貌(图3)。其中管的长度从几微米到二三十微米不等,管外径尺寸在100 nm~300 nm之间,内径约80 nm~150 nm。TEM图像更进一步证实了材料的中空管状结构,同时显示大量微小的黑色颗粒物,是有机长链阳离子与[PW9V3O40]6-阴离子组装形成的球形聚集体[18]。(DDA)6PW9V3的光学显微镜照片同样可以观察到非常清晰的中空管状结构。中空管状结构有效的增加了在反应过程中催化剂与底物的作用面积,有助于提升催化反应的速率。

图3 (a-c)不同尺寸下化合物(DDA)6PW9V3的SEM图,插图是(DDA)6PW9V3的静态接触角(134°);(d,e)不同尺寸下(DDA)6PW9V3的TEM图;(f)(DDA)6PW9V3的光学显微镜图片Fig.3 (a-c)SEM images of(DDA)6PW9V3.Inset:Static contact angle of(DDA)6PW9V3(134°);(d,e)TEM images of(DDA)6PW9V3;(f)Optical micrograph of(DDA)6PW9V3
(DA)6PW9V3、(DDA)6PW9V3、(TDA)6PW9V3、(HDA)6PW9V3、(TSA)6PW9V3和(DODA)6PW9V3的静态接触角(图S4)分别为134°、136°、121°、128°、139°和141°,表明制备的催化剂具有良好的疏水性能,有利于在低极性油品中分散,促进低极性底物在催化剂表面的传质扩散,提高底物与催化活性中心的可接近性。
2.2 催化氧化脱硫性能
2.2.1 不同催化剂萃取催化氧化脱硫效果考察
首先我们考察了不同催化剂对模拟油中DBT萃取氧化脱除效果。选取[omim]PF6离子液体为萃取剂,以H2O2为氧化剂,探索了多种类型催化剂对DBT的萃取催化氧化脱硫效果。从 表 1 可 以 看 出 ,H3PW12O40、Na8HPW9O34、Na10SiW9O34、 H6PMo9V3O40、 H5PMo10V2O40和H4PMo11VO40在60 min内对DBT催化转化效率分别为16%、48%、31%、53%、36%和30%。继续延长反应时间到6 h,对DBT催化转化率可达37%、98%、33%、89%、79%和37%。这些催化剂在常温条件下对DBT的萃取催化氧化脱硫效率较低,而Cs5PW10V2O40和K6PW9V3O40在60 min的硫去除率可达97%和99%。可以看出,含有V和W元素的杂多酸的催化效率高于其他类型杂多酸。当以(DDA)6PW9V3为催化剂时,25 min即可达到>99.9%以上的脱硫率。由此可以发现,在温和条件下,含有疏水有机官能团修饰的PW9V3催化剂更容易实现高效的萃取催化氧化脱硫。
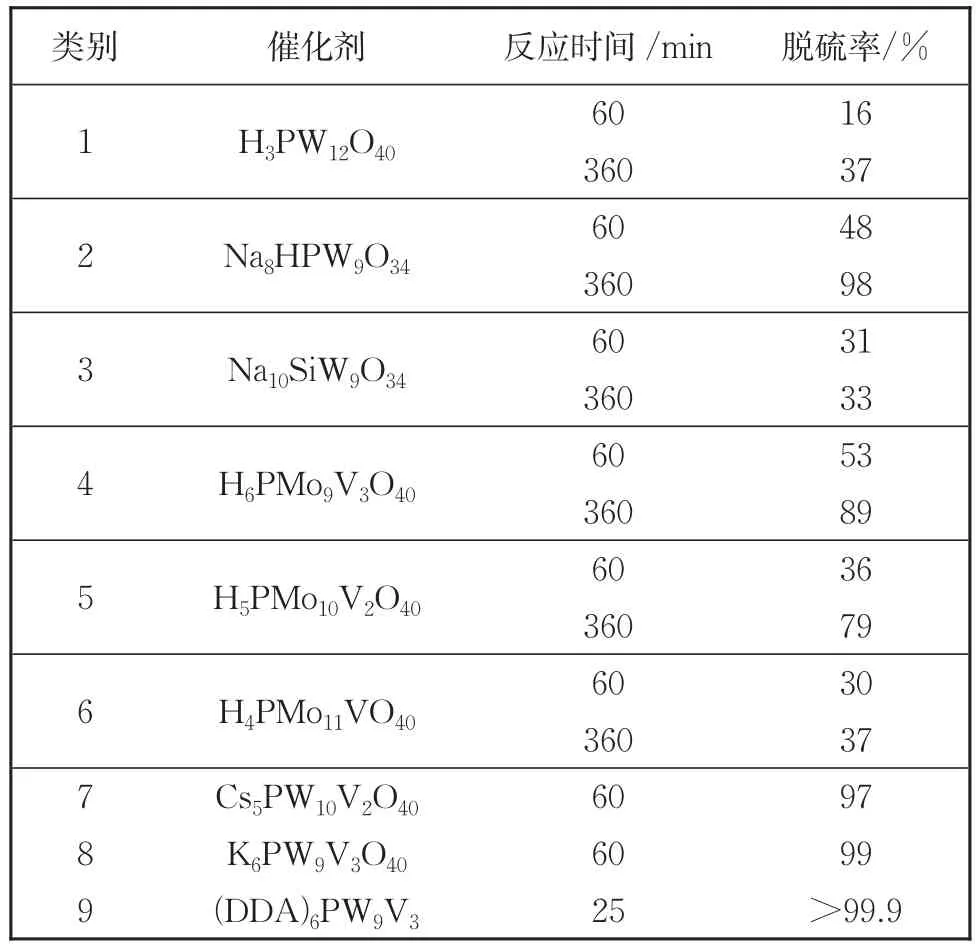
表1 不同类型催化剂萃取催化氧化脱硫效果Table 1 Extraction oxidation desulfurization effect of different catalysts
2.2.2 不同碳链长度对DBT氧化脱硫的影响规律
为了研究不同碳链长度的表面活性剂对硫脱除效率的影响,将[PW9V3O40]6-与不同烷基链长度的表面活性剂进行离子交换,得到(DA)6PW9V3、(DDA)6PW9V3、(TDA)6PW9V3、(HDA)6PW9V3、(TSA)6PW9V3和(DODA)6PW9V3催化剂。对比不同催化剂在相同条件下萃取催化氧化脱硫性能。从图4(a)可以看出:(DA)6PW9V3和(DDA)6PW9V3在30℃下25 min时DBT转化率可达99.9%(深度脱硫),30 min内使用其他类型的催化剂催化DBT的转化率分别为(TDA)6PW9V3(99%),(HDA)6PW9V3(97%),(TSA)6PW9V3(91%)以及(DODA)6PW9V3(83%)。因此,萃取催化脱硫效率对比:(DA)6PW9V3≈(DDA)6PW9V3>(TDA)6PW9V3>(HAD)6PW9V3>(TSA)6PW9V3>(DODA)6PW9V3。由此可见,表面活性剂有机链长度越短,越有利于萃取催化氧化脱硫反应。
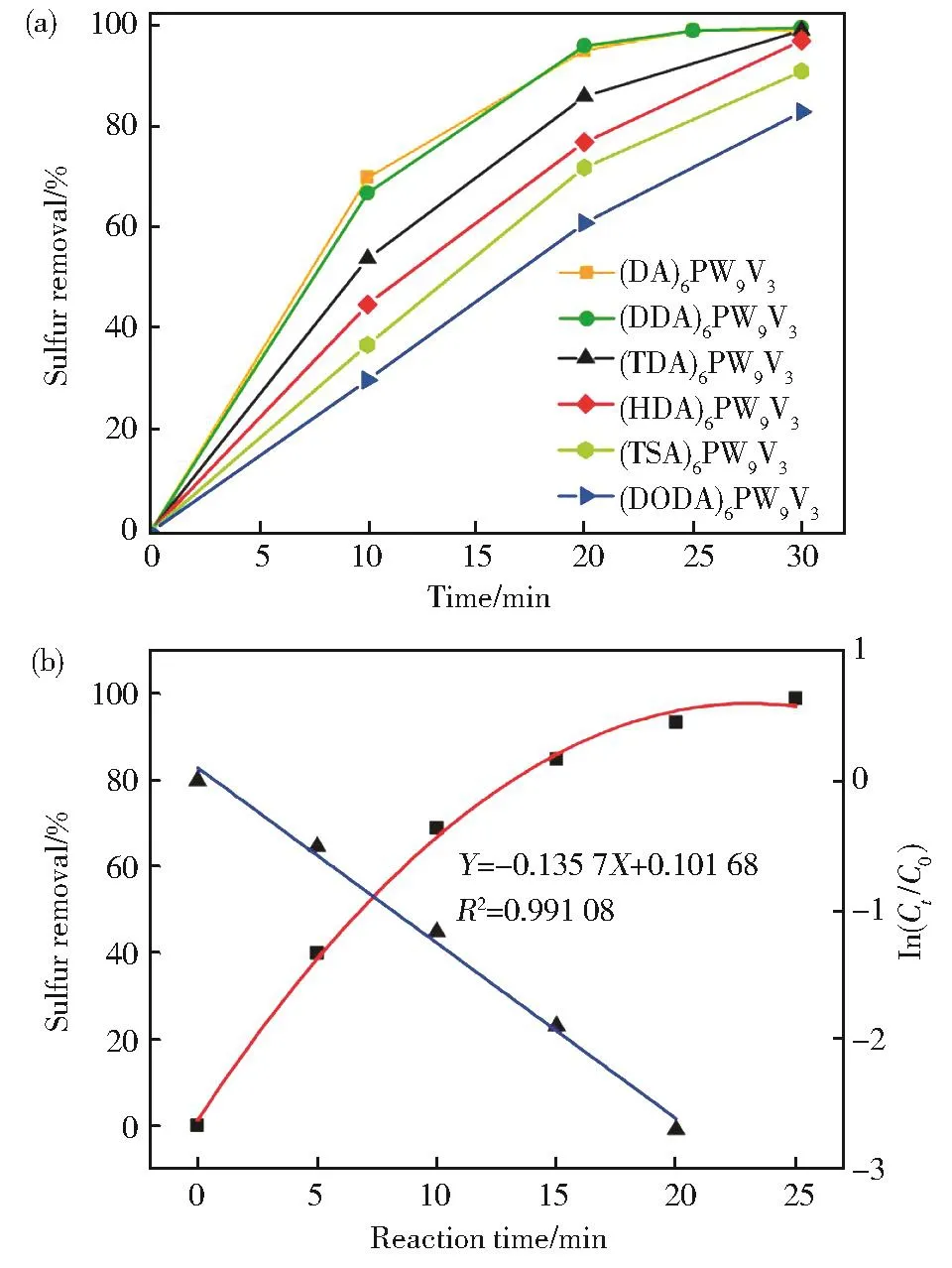
图4 (a)不同化合物在相同条件下,30 min内脱除DBT效果图;(b)DBT去除率的动力学研究曲线图,其中红色曲线代表S的去除率,蓝色直线代表ln(Ct/C0)。反应条件:H2O2∶DBT∶(DDA)6PW9V3摩尔比=250∶100∶5,硫含量1 000 μg/g,[omim]PF6用量1 mLFig.4 (a)The desulfurization of DBT within 30 minutes with different compounds under the same conditions;(b)Reaction kinetic curve of sulfur removal of DBT,and the red curve represents the sulfur removal(%),and the blue line represents ln(Ct/C0).Reaction conditions:H2O2∶DBT∶(DDA)6PW9V3molar ratio=250∶100∶5,sulfur content 1 000 μg/g,[omim]PF6:1 mL
为了得到催化氧化DBT的动力学参数,脱硫率和ln(Ct/C0)随反应时间的关系如图4(b)所示:
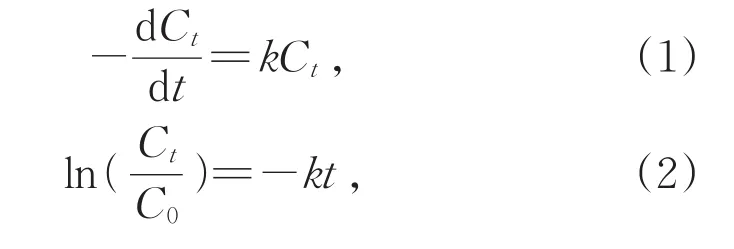
其中C0和Ct分别为DBT的初始浓度以及t时刻的浓度。数据的线性拟合表明,催化反应呈现准一级反应动力学(R2=0.991 08)。由式(1)和(2)确定了氧化反应的速率常数k为0.135 7 min-1,DBT氧化生成DBTO2的时间为25 min,DBTO2的选择性为100%,反应符合准一级动力学。最终的氧化产物经核磁共振氢谱证实为DBTO2(图 5)[16]。

图5 催化氧化脱硫前后底物DBT与生成物DBTO2的核磁共振氢谱图(氘代试剂:d3-CDCl3)Fig.5 1H NMR spectra of DBT,DBTO2(standard)and the oxidized product of DBTO2,respectively
2.2.3 不同反应温度下,(DDA)6PW9V3萃取催化氧化脱硫效果
温度对催化反应速率有着重要的影响,为了认识温度对萃取催化氧化脱硫效率的影响规律,我们选择了30℃、40℃和50℃等三个不同温度。从表2结果我们可以看出,在H2O2∶DBT∶(DDA)6PW9V3摩尔比=250∶100∶5,H2O2用量0.080 mL,模拟油用量10 mL,硫含量1 000 μg/g,[omim]PF6用量1 mL的相同反应条件下,化合物(DDA)6PW9V3萃取催化氧化脱硫效率到达99.9%所需要的时间随着反应温度的升高而变短。30℃条件下,脱硫效率达到99.9%所需要的时间为25 min,40℃时,则减少到12 min,而当反应体系的温度增加为50℃时,仅用10 min,即可实现>99.9%的脱硫效率。然而,常温下的脱硫技术能够有效减少热能的使用,是一个经济环保绿色的催化工艺,所以本篇工作重点探讨制备的系列催化剂在常温条件萃取催化氧化脱硫性能。

表2 不同反应温度下,(DDA)6PW9V3萃取催化氧化脱硫效果对比Table 2 Effect of different temperatures on the removal of DBT by(DDA)6PW9V3
2.2.4 不同 H2O2:DBT:(DDA)6PW9V3摩尔比对催化氧化脱硫影响
为了考察氧化剂和催化剂的用量对DBT萃取催化氧化脱硫性能的影响,我们设计了五种不同比例的催化反应(如表3所示)。当H2O2∶DBT∶(DDA)6PW9V3的摩尔比为 500∶100∶5时,30 min内的脱硫率>99.9%。进一步减少氧化剂的用量,当双氧水用量为 0.032 mL,即 H2O2∶DBT∶(DDA)6PW9V3的摩尔比为100∶100∶5时,30 min的脱硫效率仅为68%。而当增加催化剂用量,当使用一倍过量催化剂(H2O2∶DBT∶(DDA)6PW9V3的摩尔比为250∶100∶10)时,30 min内同样可以实现>99.9%的脱硫效率。而减少催化剂用量(H2O2∶DBT∶(DDA)6PW9V3的摩尔比为250∶100∶2),30 min的脱硫效率仅为54%。综合这些实验结果,我们选定 H2O2∶DBT∶(DDA)6PW9V3摩尔比为250∶100∶5为最优条件。
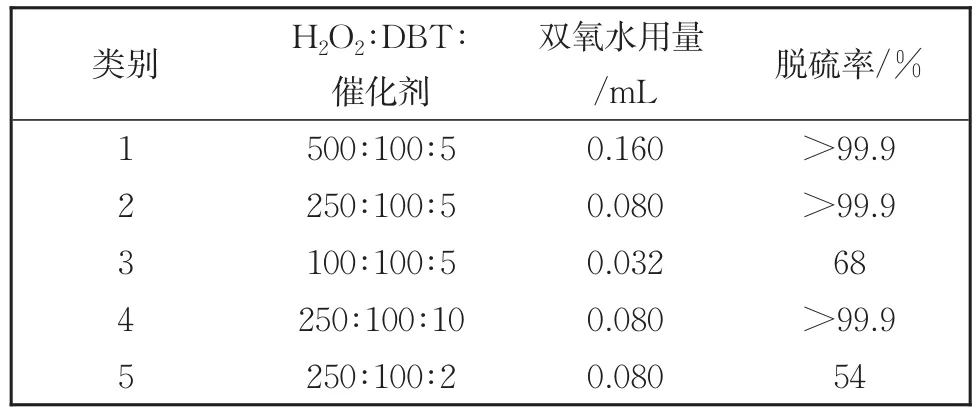
表3 不同H2O2∶DBT∶(DDA)6PW9V3比例条件下脱硫效果Table 3 Effect of different molar ratios of H2O2/DBT/Cat on the removal of DBT by(DDA)6PW9V3
2.2.5 (DDA)6PW9V3催化剂对 4,6-DMDBT 和BT脱硫效果
我们在最优反应条件下探索了(DDA)6PW9V3/[omim]PF6体系在室温条件下4,6-DMDBT和BT的萃取催化氧化脱硫反应。由于4,6-DMD⁃BT和BT存在一定的空间位阻,相对较难被氧化。如表4所示,在30℃下,在25 min内对DBT的转化率为100%,而对4,6-DMDBT和BT在60 min的脱硫率分别为86%和67%。
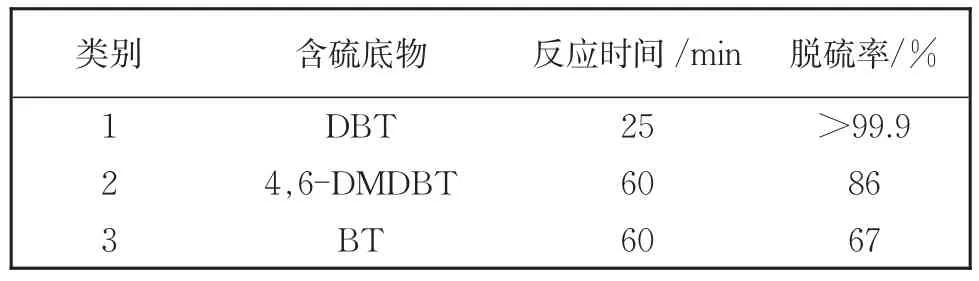
表4 (DDA)6PW9V3对DBT、4,6-DMDBT和BT的脱硫效果Table 4 Desulfuration of(DDA)6PW9V3on DBT,4,6-DMD‐BT and BT
2.3 催化剂循环利用
第一次萃取催化氧化脱硫结束后,通过离心将反应液与催化剂分离,然后将混有催化剂和离子液体的混合体系在110℃条件下干燥,以除去未反应的双氧水和水。充分干燥后,回收的催化剂在相同温度和反应时间条件下直接被用于下一次反应,重新向体系内加入新的模拟油和双氧水即可[18]。经过10次循环利用,催化剂依然保持较高的脱硫效率(图6(a)),第十次的脱硫率仍达到95.3%。进一步通过红外表征第10次循环后的(DDA)6PW9V3催化剂(图6(b)),结果显示其红外吸收的特征峰与使用前没有发生明显的变化,表明催化剂在经过多次循环使用仍能保持稳定的结构。
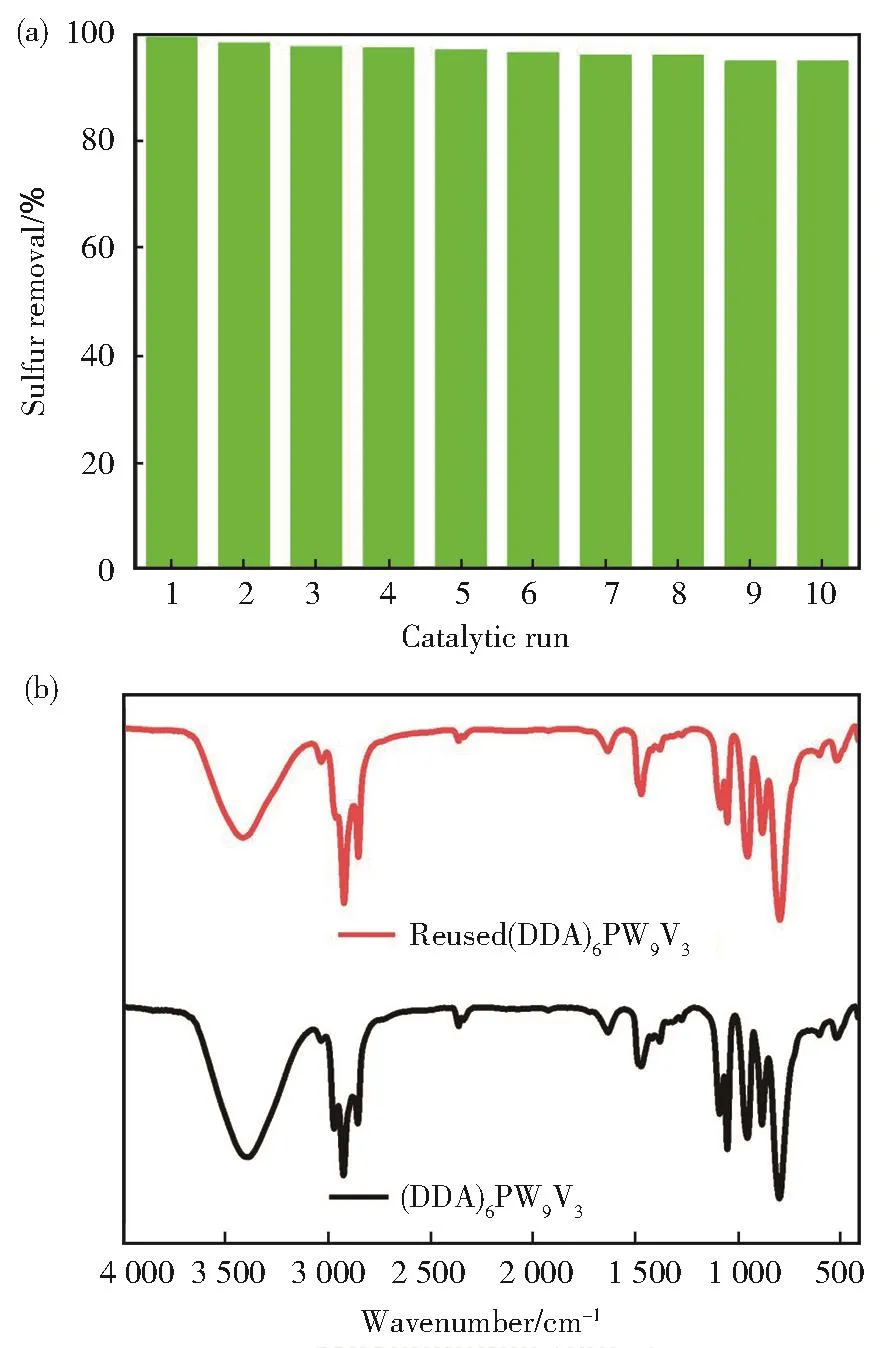
图6 (a)(DDA)6PW9V3催化剂循环10次萃取催化氧化脱硫效果图;(b)(DDA)6PW9V3催化剂使用10次前后红外谱对比Fig.6 (a)Reusability of(DDA)6PW9V3in the extraction oxi‐dation desulfurization;(b)FT-IR spectra of the fresh and used(DDA)6PW9V3
2.4 催化反应机理分析
使用(DDA)6PW9V3/[omim]PF6体系,H2O2作为氧化剂的萃取氧化催化机理与之前我们文献报道的相类似[17]。如图7所示,在搅拌过程中,油相中的底物DBT被萃取到离子液体相。催化剂的疏水长链甩在离子液体相中,低极性分子与疏水链相似相溶,促进DBT接近催化剂中心,实现了高效萃取过程。在具体氧化反应过程中,H2O2与多酸形成钨或钒的过氧自由基,将DBT分子从油相萃取到离子液体相并快速氧化成DBTO2。
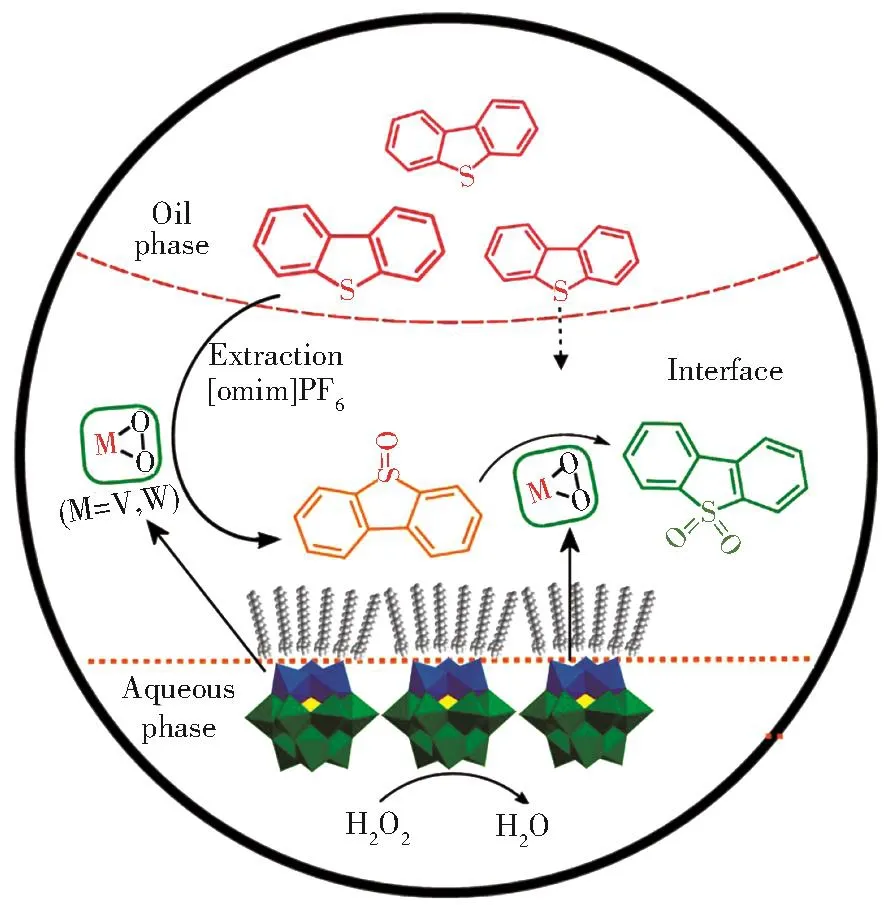
图7 (DDA)6PW9V3/[omim]PF6萃取催化氧化脱硫的反应机理Fig.7Reaction mechanism of(DDA)6PW9V3/[omim]PF6 in the extraction oxidative desulfurization
3 结论
本文采用一种简单高效的离子交换的方法成功地合成了系列具有不同长度的有机碳链修饰的含钒杂多酸。通过红外、核磁、元素分析、热重等系列表征技术,详细认识催化剂结构。在以H2O2为氧化剂,[omim]PF6为萃取剂,(DDA)6PW9V3为催化剂在30℃条件下,25 min钟内即可实现99.9%以上的脱硫率。而当温度增加到50℃时,仅需10 min即可实现99.9%深度脱硫效果。这一优异的催化效果主要归因于:(1)离子液体和疏水碳链修饰有助于提高反应底物与催化活性中心的可接近性,促进底物被催化氧化;(2)含钒杂多酸在双氧水条件下形成钨或钒的过氧自由基,对底物展现出较强的催化氧化活性,可快速氧化DBT变成DB⁃TO2。催化剂具有良好的循环稳定性,连续使用10次,脱硫效率未发生明显下降。该催化剂具有制备工艺简单、相对成本较低、反应活性高、可循环利用等特点,具有广阔的应用前景。
猜你喜欢
分子催化(2022年1期)2022-11-02
建材发展导向(2021年16期)2021-10-12
教育周报·教育论坛(2020年3期)2020-10-21
科技资讯(2018年16期)2018-10-26
科技信息·下旬刊(2018年8期)2018-10-21
智富时代(2018年3期)2018-06-11
智富时代(2018年3期)2018-06-11
中学化学(2017年6期)2017-10-16
中学化学(2017年6期)2017-10-16
科技资讯(2017年12期)2017-06-09
