
中国南方一罕见迟发性脊椎骨骺发育不良大家系的表型和基因型的相关性研究
2022-06-02 01:03郑诗瑶郭奕斌
中山大学学报(医学科学版) 2022年3期
黄 颖,谢 杰,4,郑诗瑶,唐 佳,郭奕斌,
(1.中山大学中山医学院医学遗传室,广东广州 510080;2.中山大学医学院医学遗传室,广东深圳 518107;3.广东省生殖科学研究所,广东广州 510600;4.广州盛安精准医学研究院,广东广州 510799)
迟发性脊椎骨骺发育不良(spondyloepiphyseal dysplasia tarda,SEDT,OMIM 313400)是遗传性矮小症中发病率很低的罕见遗传性骨病[1],分AR、AD、XR 遗传3 种类型,其中XR 型相对常见,患者多为男性。患病男性具有典型的躯干较短,臂展长度大于身高,且身材不成比例的矮小等症状[2],是该病临床表现中一个显著的特征。出生时,患儿的身高正常,身体比例也正常,但从6~8 岁起开始出现身高增长迟缓的现象,且逐渐矮于同龄人。影像学特征包括:不规则的扁平椎体,典型的驼峰状突起和椎间隙明显变窄[3-4]。女性携带者通常没有临床表现。Whyte 等[2]报道,女性携带者在影像学检测中仅出现细微的改变,但成年后可有轻度骨关节炎症的表现。XCI(X chromosome inactivation)是雌性哺乳动物与雄性个体之间实现基因剂量补偿的一种方式。它对许多X 连锁遗传病的发生及临床表现的严重程度有很大关系[5]。女性携带者没有SEDT 明显特征可能与此有关。本病的发生与TRAPPC2(trafficking protein particle complex subunit 2,转运蛋白粒子复合亚基2,早期称为SEDL基因的致病突变密切相关[6]。该基因定位于X 染色体的短臂(Xp22.2),gDNA 约22 kb,含有6 个外显子。编码的蛋白质含有140 个氨基酸,称为Sedlin 蛋白(SEDL 蛋白)[7]。在基因组DNA 中,有7 个已知的和SEDL高度同源的假基因(SEDLP1~SEDLP7),均不含内含子。在7 个假基因中,SEDLP1转录本的编码区与SEDL基因几乎相同,因此激活SEDLP1的表达可能成为治疗SEDT 的潜在途径[6]。SEDL 蛋白是TRAPP(运输蛋白粒子)复合物的重要组成部分,能够将原胶原三聚体(例如Ⅱ型胶原)从内质网运输到高尔基体,最终将这些蛋白质运输到细胞外基质中。它参与了细胞内物质从内质网到高尔基体间的囊泡运输的定位和膜融合的过程[8]。TRAPPC2基因的多种突变均可导致SEDL蛋白的定位错误和功能缺陷[9-10],影响其在内质网—高尔基体膜室间区的正确定位,致使细胞(尤其是软骨细胞)蛋白运输出现障碍,导致骨软骨发育不良最终引起SEDT 的发生[7,11]。本研究对来自中国南方的一遗传性矮小症家系进行详细的分子遗传学研究,最终揭示TRAPPC2基因(NM_001011658)所携带的c.94 del G,p.D32T,fsX6 突变为该家系患者发病的真正内因,为今后的产前、植入前基因诊断等创造了必要的前提条件。
1 材料与方法
1.1 病例资料
所接诊的遗传性矮小症家系来自广东四会,系谱图如图1 所示。家族中有2 名男性患者,均为其各自的母亲所遗传,符合XR 遗传规律。先证者(Ⅲ-1)是一名27岁的男子,于7~8岁时开始出现症状,现身高140 cm,臂展长度为151 cm。颈部短,劳累时背部出现明显疼痛,髋部和肩部也出现轻度疼痛。膝关节尚无症状,鸡胸,头围正常,无异常的面部特征,智力、外耳、心肺均正常。先证者的舅舅(Ⅱ-3)与先证者有相似的临床症状和体征,身高148 cm,臂展长度为157 cm。先证者的母亲(Ⅱ-2)共四姐弟,在确定先证者(Ⅲ-1)及其舅舅(Ⅱ-3)为患者的前提下,根据遗传规律,推测系谱中的4 人即先证者的外婆(Ⅰ-2)、母亲(Ⅱ-2)、表妹(Ⅲ-4)及女儿(Ⅳ-1)应为携带者(图1),后经测序得到验证。家族中表型正常的男性和正常对照组均无此突变。

图1 广东一矮小症家系第一次遗传咨询系谱图Fig.1 The first genetic counseling pedigree of a dwarfism family from Guangdong
为了进一步明确基因型与表型的相关性,我们采集了家族中更多的成员资料和血样进行测序验证,系谱图见图2。本研究是在患者及其家属签署知情同意书的情况下开展的,并获得了中山大学医学院医学伦理委员会的批准(批准号:医学院医伦[2021]第57号)。

图2 广东一矮小症大家系的扩大系谱图Fig.2 Extended pedigree of a dwarfism large family from Guangdong
1.2 方 法
1.2.1 全外显子组测序 采集先证者(Ⅲ-1)及其妻子(Ⅲ-2)、女儿(Ⅳ-1)的血样,提取DNA,测定DNA 纯度和浓度,进行全外显子组测序(iGeneTech,Beijing,China),重点检测跟遗传性矮小症相关的基因。超过90%的目标区域是用覆盖≥30X 的深度测序,通过使用带有标准参数的Burrows-Wheeler aligner(BWA)将测序原始数据映射到UCSC 数据库人类参考基因组(版本:hg19)上进行比对[12]。重复的读数由Picard(http://Broadstitute.github.io/picard/)标记,通过使用基因组分析工具包(GATK)[13]和最佳实践推荐参数进行区域比对和质量评分重新校准,以调用单核苷酸变体(SNV)和短插入/缺失变体(INDELs)。使用软件平台KGGSeq(http://pmglab.top/kggseq/)[14-15]对候选突变进行三个层次(遗传水平、变体-基因水平和知识水平)的筛选和注释,最后通过表型数据库和文献检索,筛选出与遗传性矮小症或表型相关的候选突变。
1.2.2 Sanger 测序验证TRAPPC2基因突变 采集全部受试者的外周血,提取基因组DNA,采用Primer Premier 5.0 设计引物(参考序列NG_011555)(表1)。按以下条件扩增TRAPPC2基因的四个外显子和与其相邻的内含子序列:95 ℃预变性5 min,然后94 ℃变性30 s,58 ℃退火40 s,72 ℃延伸50 s,循环次数为35 次,72 ℃再延伸3 min。采用琼脂糖凝胶电泳检测扩增产物,并对扩增产物采用ABI PRISM 3730 型测序仪进行双向测序(上海Invitrogen 生物技术公司)。测序结果由Chromas 软件读取,并与参考序列进行比较分析。
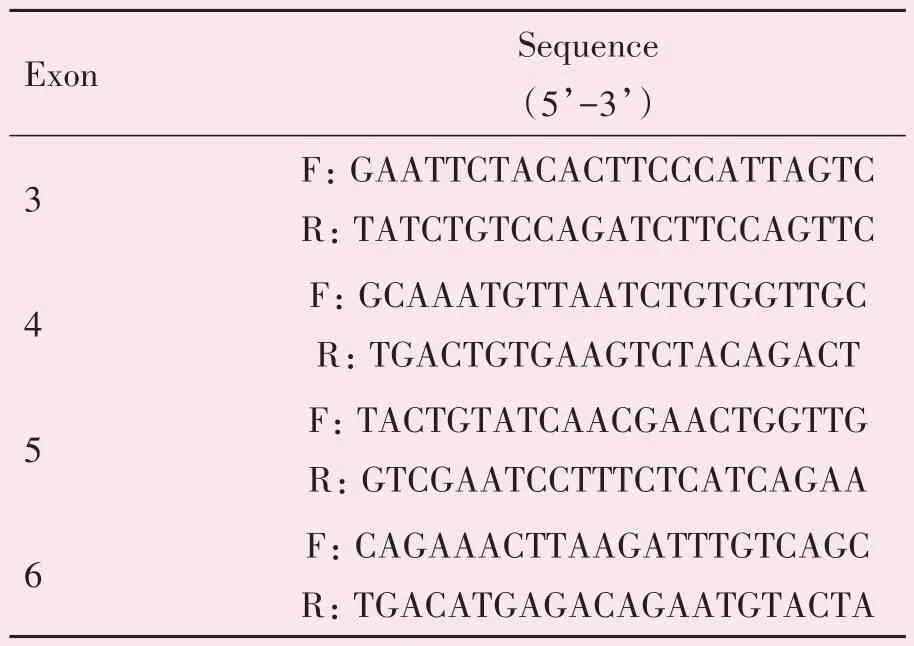
表1 TRAPPC2基因PCR引物序列Table 1 PCR primer sequences for TRAPPC2
1.2.3 逆转录PCR(RT-PCR)使用Trizol 试剂(Simgen试剂盒),从受试者新鲜外周血白细胞中提取总RNA,在20µL 体积内反向转录1 000 ng RNA(Fermantas 试剂盒)。选用cDNA 作为逆转录PCR的反应模板,逆转录PCR 的引物为:F:5’-GCGCAAGCTTGCCATATATTGAAGACCATGTCTGGG -3’;R:5’-GCGCGGATCCCGAGTATACACCATTGTGGTGACATC -3’。PCR 反应条件同1.2.2 对逆转录PCR 产物进行双向测序(上海Invitgen 生物技术公司)。
1.2.4 实时荧光定量PCR(qPCR)采用TRIzol法提取全部受试者的外周血中总RNA,设计了跨越3 号和4 号外显子的引物(E3F:5’-GAGCTTCTACTTTGTAATTGTTGGC -3’;E4R:5’-AGTGACAAATGCCGACACAAA -3’)。由于TRAPPC2基因与假基因SEDLP1的同源性高达98%[6],本研究将引物3’端设计在两个基因的错配区,以保证只有TRAPPC2基因能扩增出来。以β-actin 为内部参照。反应总体积为20µL,包括稀释的cDNA、引物和SYBR Green I Master Mix(Fermentas k0222 试剂盒)。反应条件为95 ℃预变性10 min,95 ℃变性15 s,62 ℃退火60 s,95 ℃变性15 s,然后60 ℃延伸60 s,95 ℃变性15 s。每个样品重复检测3 次,应用比较CT 法进行分析数据。
1.2.5 变异分析与致病性评估 经检索HGMD(人类基因突变数据库,http://www.hgmd.cf.ac.uk/ac/index.php)、dbSNP(NCBI,http://www.ncbi.nlm.nih.gov/snp)和千人基因组计划数据库,确认我们所发现的TRAPPC2基因的c.94 del G,p.D32T,fsX6 新变异还未被报道。然后,使用Clustal Omega 在11 个SEDL 蛋白质同源物的fasta 格式文件上执行多序列比对[16]。此外,还使用SWISSMODEL[17](https://swissmodel.expasy.org/)对野生型和突变型SEDL 蛋白的三级结构进行了比对,PYMOL 软件用于可视化蛋白质的三级结构的分析,并标出突变位置。结合临床资料、分子遗传学检测结果和新变异体的一系列鉴定结果,我们参照ACMG指南[18-19]综合分析和评估TRAPPC2基因中的新变异的致病性。
2 结果
2.1 全外测序结果
首先通过计算亲缘系数,排除先证者父母三代内近亲婚配的可能性。然后通过对变异的过滤注释和质控,包括过滤掉内含子区的变异和同义突变,保留次要等位基因频率(MAF)<3%或不在突变数据库中,以及相符的遗传模式过滤,筛选与疾病相关的候选变异。使用KGGSeq 和VarCards 对变异的有害性进行评估,通过对候选变异相应基因的疾病种类和表型与先证者家系疾病表型的相似性进行整合分析,最终位于TRAPPC2基因(NM_001011658)的单碱基缺失变异c.94 del G,p.D32T,fsX6 被选为候选突变,进行Sanger 测序验证。
2.2 Sanger测序验证结果
经Sanger 测序验证,该家系的先证者及家族中的所有患者和肯定携带者均被证实存在上述变异(缺失单碱基G),先证者和其他患者是半合子,先证者的女儿和母亲都是杂合子,而正常对照组无此变异(图3A)。由于3 号内含子的最后一个碱基和4 号外显子的第一个碱基都是G,所以在DNA水平无法确定缺失的确切位置。因为如果缺失的G 位于3 号内含子的最后一个位点,则突变应为IVS3 as(-1)del G,但如果缺失的G 是4 号外显子中的第一个碱基,则突变为c.94 del G。确切的变异位点需用逆转录PCR(RT-PCR)在RNA 水平上来鉴定。通过采集该家系更多成员的血样进行测序验证,结果与理论预测的完全相符。该家族健在的女性有12 人,其中肯定携带者6 人,可能携带者6 人。男性患者4 人。在可能携带者(图1 打“?”者)中,检出携带致病基因的有4 人,分别是:Ⅳ-5、Ⅳ-7、Ⅴ-3、Ⅴ-7。另外,Ⅴ-6 未检,Ⅴ-9 正常(图2)。
2.3 RT-PCR鉴定结果
在该家系中,RNA 样本被逆转录成cDNA,作为RT-PCR 的反应模板。测序结果显示,患者的转录本中的G 碱基被删除,并且剪接位点没有发生变化(图3B)。因此,通过RT-PCR 可以证实,该家系的变异是由于外显子4 的第一个碱基G 的缺失引起的移码突变。c.94 del G 导致TRAPPC2 蛋白第32 位的氨基酸由D 变为T,并在产生5 个新的氨基酸后,出现了一个TGA 终止密码子(c.94 del G,p.D32T,fsX6),从而导致TRAPPC2 蛋白从140 个氨基酸截断至36个氨基酸。

图3 TRAPPC2基因Sanger测序的结果Fig.3 The results of Sanger sequencing for TRAPPC2
2.4 荧光定量PCR检测结果
对正常组(Normal)、杂合子携带者组(Carriers)和半合子患者组(Patients)的TRAPPC2基因的RNA 的相对表达量进行分析,三个不同分组的TRAPPC2基因相对表达量的分析结果如图4所示。以正常对照组(Normal)的“1.000”为单位,相对于β-actin 分析的表达水平,3 个组的TRAPPC2基因的相对表达量分别为1.000(Normal)、0.561(Carriers)和0.489(Patients)(图4)。经单因素方差分析,三组间差异有统计学意义(F=38.31,P=0.000 4);采用Bonferroni 法作两两比较,发现患者组与携带者组比较差异无统计学意义(P=0.909 6),而患者组与正常对照组比较差异有统计学意义(P=0.000 6),且携带者组与正常对照组比较差异有统计学意义(P=0.001 3)。分析表明,患者和携带者TRAPPC2基因的表达水平均明显降低,且患者的表达水平比携带者减少得更多。

图4 TRAPPC2 基因在正常、携带者和患者组中的相对表达水平Fig.4 Relative expression levels of TRAPPC2 gene in Normal,Carrier and Patient group
2.5 致病性鉴定结果
应用序列比对在线软件CLUSTAL Omega 对11个跨物种的TRAPPC2 蛋白质的氨基酸序列进行分析、比对。不同的物种包括:非洲爪蛙(Xenopus)、斑马鱼(Danio)、原鸡(Gallus)、褐鼠(Rattus)、野猪(Sus)、牛(Bos)、人(Homo)、类人猿(Pongo)、黑猩猩(Pan)、恒河猴(Macaca)、雪豹(Mustela)。物种保守性分析结果显示,p.32D(天冬氨酸)在11 个跨物种中均高度保守,且后续的氨基酸序列也都高度保守(图5)。提示该区域氨基酸序列可能具有重要的生化功能,新突变所带来的氨基酸的变化可能会破坏该蛋白质的正常功能。c.94 del G 突变导致TRAPPC2蛋白从第32个氨基酸开始产生几个新的氨基酸,导致蛋白质截短后产生了一个新的终止密码子。这些变化无疑会破坏正常TRAPPC2 蛋白的原有功能,从改变蛋白的一级结构到最终影响、破坏蛋白的生化或生理功能。

图5 TRAPPC2基因的CLUSTAL Omega分析Fig.5 CLUSTAL Omega analysis of TRAPPC2
通过SWISS-MODEL 预测TRAPPC2 蛋白的三级结构,我们发现单碱基缺失突变(c.94 del G,p.D32T,fsX6)引起移码并导致翻译提前终止,肽链明显截短,从而导致sedlin 的功能受到严重干扰。与野生型TRAPPC2 蛋白(图6A)相比,突变蛋白的三级结构发生了明显改变(图6B)。根据ACMG 指南[18-19]对新变异的致病性进行评估,该变异属于移码突变,与ACMG 指南中对PVS1(非常强的致病性证据)的描述一致。其次,在数据库群体排查中未发现该变异,与PM2_Supporting 的描述一致。第三,支持证据PP1、PP3 和PP4 的描述也与该变异一致。综上所述,该新变异包括1个非常强(PVS1)和4 个支持性证据(PM2_Supporting,PP1,PP3,PP4)结合在一起。因此,综合上述所有分析和实验结果,该家系的新变异被鉴定为致病性突变。
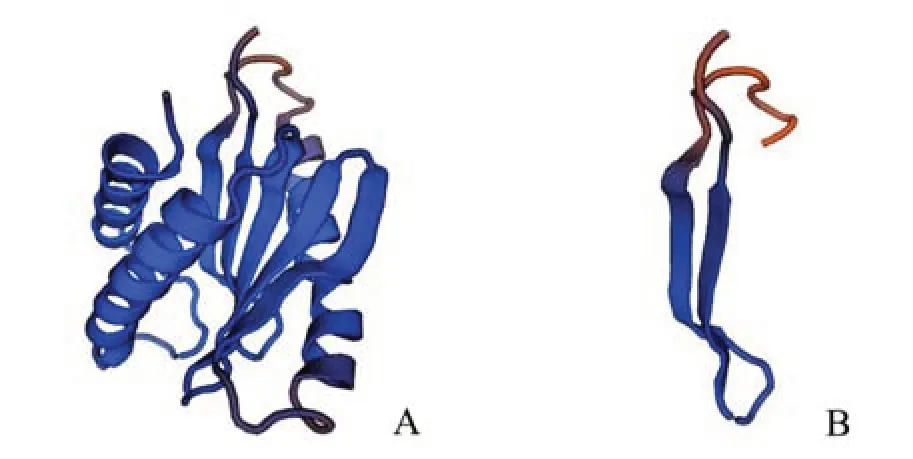
图6 TRAPPC2蛋白的突变型和野生型三级结构Fig.6 The mutant type and wild type tertiary structures of TRAPPC2 protein
3 讨论
遗传性骨病种类多,危害严重,再发风险高,但发病率低,病因复杂,尤其是罕见疑难骨病。因此,揭示其分子发病机制,对高危家庭通过产前/植入前诊断实现优生具有重要意义。由于遗传性骨病具有高度的遗传异质性和表型多样性,加上刚接诊时,患者症状不是特别典型,家族成员的资料也不多,所以为了尽快查清病因,我们直接采用NGS 测序技术对与矮小症相关的基因包进行了比较全面的筛检,重点筛检与骨骼系统相关的基因包括TRIP11、COL2A1、FGFR3、ARSB、PTH1、IMPAD1、TRAPPC2、EXT1、PDE4D、PRKAR1A、GDF5、NOG等100 多个。经临床表型分析、系谱分析、生物信息分析、实验验证,最终确定TRAPPC2基因(NM_001011658)所携带的c.94 del G,p.D32T,fsX6 突变为该家系患者发病的真正内因,所患疾病为迟发性脊椎骨骺发育不良(SEDT)。
SEDT 是一种骨软骨发育不良的疾病,其特征在于脊柱骨的短缩,长骨骨骺端的不规则变化。该病具有高度的表型异质性[20],很容易与其他骨病相互混淆且由于发病率很低,临床医生对该疾病的认识也不够全面,TRAPPC2基因的致病性突变是绝大多数SEDT 发病的根本原因。TRAPPC2 蛋白在进化过程中高度保守,在脑、心、肾、肝、肺、胰腺、胎盘、骨骼肌、胎儿软骨、纤维细胞和淋巴细胞中均有表达[20]。以往对酵母同源基因p20 的研究发现,sedlin 蛋白构成TRAPP(运输蛋白颗粒)的亚基,参与内质网(ER)的靶向和融合[21]。TRAPPC2基因突变可能导致蛋白的定位错误和功能缺陷,导致软骨细胞蛋白复合体从内质网转运到高尔基体的障碍[9-10]。根据HGMD 和相应的文献报道[22],迄今为止已有57 种不同的突变类型,包括无义突变、错义突变、缺失、插入和剪切位点等突变类型。值得注意的是,三号内含子和四号内含子下游,很可能影响剪切位点的变异至今还没有被发现或鉴定。还有文献指出,TRAPPC2基因的突变距离5’端越近,其临床表型将越严重[7]。本研究发现的移码突变(p.D32T,fsX6)是由外显子4 的第一个碱基G 缺失引起的,至今仍未见报道,经本研究详细的分析和致病性鉴定,证实为一种新的致病突变,从而丰富了TRAPPC2 基因的突变谱。通过本论文研究还得到启发,对发生在外显子和内含子交界部位的单碱基新变异,通过Sanger 测序和RT-PCR、q-PCR 检测,从DNA、RNA 等多个水平对新变异进行鉴定是很有必要的。
另据文献报道,人类基因组中至少有7 个TRAPPC2假基因[23]。SEDLP1位于染色体19q13.4,SEDLP2位于染色体8q13.3,SEDLP3-SEDLP7位于染色体Y q11.23.21。在TRAPPC2基因和SEDLP1假基因的开放阅读框上,仅有6 个同义突变核苷酸存在差异。已证实SEDLP1在组织中转录并广泛表达,并编码一种蛋白,与TRAPPC2基因高度同源[6]。因此,设计用于扩增全长TRAPPC2基因和Real-Time PCR 的引物时,应避免SEDLP1的扩增。根据文献[23]推测,SEDLP1基因导入细胞后可能在细胞核周围结构中表达,我们推测由于某种补偿机制,SEDLP1在女性携带者在一条X 染色体上发生基因突变的情况下表达取代了TRAPPC2的功能,从而导致TRAPPC2 蛋白表达下降。另一方面,TRAPPC2基因被证实逃脱了X 染色体失活(XCI)。XCI 是雌性哺乳动物和雄性哺乳动物之间实现基因剂量补偿的一种方式,这与临床表现的严重程度有关[5]。一些研究阐明,雌性携带者的野生型和突变型可同时表达,由此说明TRAPPC2基因逃脱了X染色体的失活,但Whyte等[2]报道,成年女性携带者的影像检查只是轻微的异常,甚至有些人只是患有骨关节炎。
综上,SEDT的潜在并发症是早发性关节炎,表现为关节痛和活动受限。对于病情严重的患者,应在成年初期实施髋部正畸治疗[24]。目前对SEDT尚无有效的治疗方法,但是,当SEDT患者的骨骼在婴幼儿和儿童早期发育完全正常时,采取基因干预治疗可能是可行的[25],但基因治疗方法目前仍处于实验阶段。因此,加强预防工作显得更为重要。由于该病发病年龄较晚,产前B 超检查不能确诊,因此,在明确先证者病因的前提下,对胎儿进行产前或植入前基因诊断,从而防止患胎出生、真正实现优生是目前最有效的诊防策略。SEDT患者的基因诊断不仅有利于临床诊断,也有利于女性携带者的诊断,也为预测复发风险和遗传咨询奠定了基础。这正是本研究的意义之所在。
猜你喜欢
分子诊断与治疗杂志(2022年9期)2022-10-09
科学之谜(2021年2期)2021-04-25
幸福家庭(2020年17期)2020-12-10
科学导报(2020年54期)2020-09-09
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
家庭科学·新健康(2017年5期)2017-05-18
故事作文·高年级(2017年3期)2017-04-12
家庭科学·新健康(2016年5期)2016-05-12
湖北农业科学(2014年11期)2014-09-10
