
NADPH氧化酶4在急性肾损伤中的作用机制
2022-05-18 02:28杨乐天综述赵宇亮审校
肾脏病与透析肾移植杂志 2022年2期
李 健 杨乐天,2 综述 赵宇亮,2 审校
急性肾损伤(AKI)表现为短时间内肾功能快速下降,发病率和重症患者死亡率均较高,部分AKI患者肾功能无法恢复,可进展至慢性肾脏病甚至终末期肾病[1]。AKI常见病因包括缺血/再灌注损伤(IRI)、脓毒症及肾毒性药物(如顺铂)等,其发病机制复杂多样,氧化应激、炎症、免疫损伤等因素均可能参与其中。目前AKI患者的管理以对症为主,临床上尚缺乏针对性的治疗药物。既往研究表明活性氧(ROS)与AKI密切相关,ROS含量失调可引起氧化应激,触发细胞凋亡或坏死,最终引起AKI[2]。还原型烟酰胺腺嘌呤二核苷酸磷酸(NADPH)氧化酶4(NOX4)是体内ROS重要来源之一,在各型AKI中出现异常表达,可能在其发病机制中发挥了重要作用,而以NOX4为治疗靶点的干预策略也逐渐受到学界的重视。本文总结NOX4在AKI中的作用机制及其应用前景,为探究AKI新型治疗靶点提供思路。
NOX家族
NOX家族是通过生物膜向氧传递电子生成超氧化物的一种蛋白质类的酶[3],由NOX1、 NOX2、NOX3、NOX4、NOX5、DUOX1和DUOX2七种亚型组成[4]。其中NOX4在肾脏中高表达,通过催化亚基促使电子从NADPH转移到氧分子上形成过氧化氢(H2O2),是组织细胞 ROS的主要来源之一,在糖尿病肾病、肾癌、AKI等多种肾脏疾病发挥着至关重要的作用,因此我们重点关注NOX4,并对其定位、性质和作用进行总结。Geiszt等[5]通过原位杂交实验确定了在小鼠肾脏中NOX4 mRNA定位于肾皮质,且主要位于近端小管的上皮细胞;而在人类肾脏中发现NOX4 mRNA主要位于髓质集合管和肾乳头上皮中。Gorin等[6]发现肾小管系膜细胞中也较低水平表达NOX4。ROS的主要作用包括细胞防御、激素合成、信号转导、G蛋白耦联受体活化、调节离子通道的活性等,同时也参与转录因子和基因表达的调节。当组织中ROS过量时,又会导致氧化应激,通过激活丝裂原活化蛋白激酶(MAPK)等信号通路,诱导细胞凋亡,造成严重的病理损伤[4],是导致AKI发生发展的重要机制。
NOX4与AKI
NOX4与许多肾脏疾病有密切的关系。糖尿病肾病中系膜细胞和足细胞中NOX4表达增加,使得ROS生成增多,进而导致氧化应激损伤,是糖尿病肾病发生和发展的重要机制之一[7]。在梗阻性肾病中,如小鼠单侧输尿管梗阻(UUO)模型中,NOX4诱导ROS的产生,引起线粒体损伤和肾实质细胞凋亡,也有研究显示在UUO模型中NOX4在损伤的肾小管中表达减少[8]。NOX4还通过激活下游信号通路介导血管紧张素Ⅱ从而诱导的肾脏损伤[9],在高血压肾病的发生发展中发挥重要作用。同时NOX4也是肾癌细胞ROS的主要来源,通过促进低氧诱导因子2(HIF-2)的表达在肾肿瘤中发挥关键作用[10]。以下针对NOX4在各型AKI中的作用机制进行阐述。
脓毒症AKI(SAKI)脓毒症是机体对感染反应失调引起的危及生命的器官功能障碍。肾脏是其中最容易累及的器官之一,易导致SAKI[11]。SAKI的病理生理机制很复杂,目前尚未完全明确,主要包括凝血功能紊乱,炎症因子风暴,小管上皮细胞能量代谢障碍等。
研究认为SAKI时NOX4过度激活产生大量ROS,引起肾小管上皮细胞氧化应激与线粒体功能障碍,从而导致细胞凋亡坏死和SAKI[3]。Sureshbabu等[12]的研究中利用脂多糖(LPS)刺激人肾近端肾小管上皮细胞(HK-2)建立SAKI肾小管损伤的体外模型,以盲肠结扎穿刺(CLP)建立体内模型。实验显示,模型组HK-2细胞和小鼠肾脏组织磷酸化受体相互作用蛋白激酶3(p-RIPK3)和磷酸化混合系列蛋白激酶样结构域(p-MLKL)的表达水平较对照组均增加,继而通过转录后机制上调NOX4表达和线粒体易位,最终导致氧化应激和线粒体功能障碍,诱发SAKI。
Yoo等[13]发现含 Ysc84 样1的Src 同源 3 域(SH3YL1)中的SYLF结构域可与NOX4的NADPH结合位点相互作用。LPS可诱导肾小管上皮细胞中的内源性NOX4高表达,与SH3YL1和p22phox结合成为三元复合物,其中SH3YL1充当衔接蛋白。当在小鼠腹腔注射LPS后,首先作用于Toll样受体(TLR),激活下游级联激酶,如脾酪氨酸激酶(Syk)等。Bae等[14]也指出Syk在TLR4信号转导中发挥了重要作用,使得SH3YL1的酪氨酸位点磷酸化,进一步形成三元复合物,激活后续通路。TLR4-SH3YL1-NOX4级联在LPS诱导的肾损伤中起重要作用。Park等[15]的研究指出了NOX4末端的COOH末端区域参与了LPS介导的核因子κB(NF-κB)的活化。该通路活化可生成H2O2,活化NF-κB,生成促炎因子[白细胞介素1β(IL-1β)和肿瘤坏死因子α(TNF-α)]等,也有研究表明[16],巨噬细胞中的核苷酸结合结构域样受体蛋白3(NLRP3)也通过此通路活化,NLRP3激活的第一阶段由TLR4受体诱导,使得NF-κB通路活化,启动IL-1β,IL-18和NLRP3的转录和翻译,第二阶段ROS等刺激信号与NLRP3受体结合,自我激活水解产生含半胱氨酸的天冬氨酸蛋白水解酶1(caspase-1),这是引起炎症的主要原因[16],从而使得肾脏中的巨噬细胞和中性粒细胞增多,并介导氧化应激、炎症、细胞凋亡等,最终引起SAKI(图1)。

图1 脓毒症AKI的NOX4相关通路示意图NOX4:NADPH氧化酶4;AKI:急性肾损伤;RIPK3:受体相互作用蛋白激酶3;MLKL:混合系列蛋白激酶样结构域;SH3YL1:含 Ysc84 样1的Src 同源 3 域;TLR: Toll样受体;Syk:脾酪氨酸激酶;NLRP3:核苷酸结合结构域样受体蛋白3;NF-κB:核因子κB;IL-1β:白细胞介素1β;caspase-1:含半胱氨酸的天冬氨酸蛋白水解酶1
顺铂相关AKI顺铂是一种经典的化疗剂,可用于多种肿瘤的治疗,但由于存在骨髓抑制、肾毒性和耳毒性等副作用,其使用常受限[17]。ROS导致的肾小管上皮细胞(RTECs)损伤是顺铂诱导AKI的主要原因[18]。Dutta等[19]研究发现在顺铂诱导的AKI中,顺铂会激活肌醇加氧酶(MIOX),从而激活NOX4并引起后续的反应。而在另一个研究中提出[20],当顺铂诱导时,原本在肾小管内高表达的硒蛋白T(SelT)表达减少,促使NOX4的活化,促进了ROS的积累。 Meng等[21]的体内外实验中均发现,过表达NOX4后顺铂诱导的肾损伤分子1(KIM-1)上调,炎症细胞因子TNF-α,IL-1β和IL-6的表达水平均增加,从而加速了顺铂诱导的AKI;在体外实验中过表达NOX4的HK-2细胞中,RIP1/RIP3/MLKL轴被激活,并促进了Caspase-3的裂解,导致ROS大量产生,最终诱发AKI(图2)。

图2 顺铂相关AKI的NOX4相关通路示意图NOX4:NADPH氧化酶4;AKI:急性肾损伤;PA:原儿茶醛;DEX:右美托咪啶;DIM:二吲哚基甲烷;MIOX:肌醇加氧酶;SelT:硒蛋白T;ROS:活性氧簇;KIM-1:肾损伤分子1;IL-1β:白细胞介素1β;TNF-α:肿瘤坏死因子α;caspase-3:含半胱氨酸的天冬氨酸蛋白水解酶3
IRI相关AKIIRI是AKI常见的原因之一,在肾移植过程中不可避免,可能激活固有免疫而引起炎症和细胞损伤。目前已经有研究表明低氧会造成转化生长因子(TGF-β1)的上调,Cho等[22]进一步研究证明了TGF-β1/Smad/NOX4途径激活细胞凋亡。除Smad通路外,TGF-β1还通过PI3K通路、 MAPK通路和RhoA/ROCK通路上调NOX4的表达[23]。Li等[24]进一步研究发现心肌素相关转录因子A(MRTF-A)通过与乙酰转移酶MYST1相互作用促进H4K16乙酰化,并激活NOX4的转录,表达NOX4。Ben Mkaddem等[25]通过夹闭小鼠双侧肾蒂模拟IRI,发现低氧条件会刺激NOX4和TLR4相互作用,在低氧诱导的内质网激活凋亡信号调节激酶1/c-Jun氨基末端激酶(ASK1/JNK)通路,即Cho等[22]提出的丝裂原活化蛋白激酶(MAPKs) 途径中发挥重要作用,从而增加p-JNK和p-p38的含量,产生ROS,导致AKI。2018年Jeong等[26]的研究也证明了MAPKs通路激活后产生的ROS在IRI-AKI细胞凋亡中扮演了重要作用。
2016年的一项研究则认为NOX4在IRI中发挥保护作用[27]。在该研究中NOX4敲除小鼠较野生型小鼠肾功能下降,肾小管凋亡加重,Bcl-2表达降低,核因子2(NRF2)活性降低。体外试验中沉默NOX4细胞具有明显的凋亡倾向,NRF2、谷胱甘肽含量和Bcl-2表达降低。NRF2是一种转录因子,在防止过度氧化中发挥重要作用,Bcl2是一种抗凋亡蛋白,NRF2是一种转录因子,NOX4可能通过改变Kelch样ECH相关蛋白1(Keap1)的氧化状态来调节NRF2的活性,继而影响谷胱甘肽的合成和氧化应激水平[28]。同时也有研究表明NOX4通过调节真核起始因子2(eIF2)的亚基eIF2α介导的信号传导保护应激条件下的细胞,从而保护肾小管细胞免受内质网应激介导的凋亡[29]。这些研究提示的NOX4保护性作用与其他研究所认为的NOX4在AKI扮演的致病作用有很大的不同。NOX4对AKI的保护或损伤作用有可能取决于不同细胞和不同情况下需要ROS量的差异(图3)。
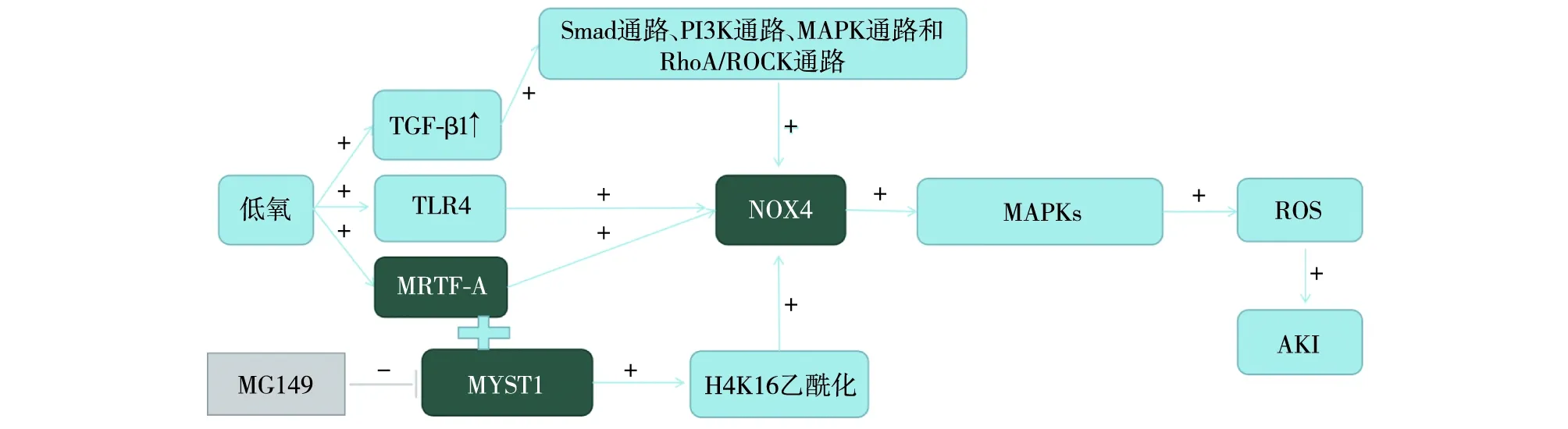
图3 IRI相关AKI的NOX4相关通路示意图IRI:缺血/再灌注损伤;NOX4:NADPH氧化酶4;AKI:急性肾损伤;MRTF-A:肌素相关转录因子A;MAPKs:丝裂原活化蛋白激酶;MYST1:乙酰转移酶;TGF-β1:转化生长因子;ROS:活性氧簇;TLR4:Toll样受体4;MG149:2-[2-(4-庚基苯基)乙基]-6-羟基苯甲酸;MRTD-A与MYST1相互作用促进H4K16乙酰化,并促进NOX4的表达
潜在干预靶点
直接干预NOX4体外试验中原儿茶醛(PA)可靶向作用于NOX4,抑制RIP1/RIP3/MLKL轴,显著降低了顺铂诱导的HK2细胞中ROS的产生,从而减少炎症和细胞凋亡,对AKI有一定的缓解作用[30](图1、2)。
右美托咪啶(dexmedetomidine,DEX)在用LPS构建的大鼠体内试验过程中发现可抑制TLR4、NOX4、NF-κB和NLRP3等的表达,即通过抑制TLR4/NOX4/NF-κB途径,减轻炎症和氧化应激,从而缓解脓毒性AKI[31]。迄今共有10项使用DEX治疗AKI临床试验注册,尚未见相关试验结果的文献报道。
硒蛋白T(SelT)可能作为新的靶点,降低NOX4和ROS的含量,抑制氧化应激和细胞凋亡,在顺铂诱导的AKI中发挥重要作用,但具体机制尚未明确[20]。
胆钙化醇是维生素D3的一种非活性形式,在缺血/再灌注诱导的小鼠AKI体内实验模型中用胆钙化醇预处理可抑制NOX的亚基p47phox和gp91phox,从而减轻肾脏病理损伤和AKI[32]。体内、外试验中杜鹃素均可抑制MAPK途径,并下调p-NF-κB和NLRP3蛋白质表达以抑制炎症,抑制NOX4的产生,同时杜鹃素还可通过激活NRF2抑制炎症和氧化应激从而对AKI 有一定的治疗效果[33]。在AKI小鼠模型中3,3‘-二吲哚基甲烷可有效地阻断LPS诱导的NOX4和NOX2的表达,从而减少ROS的产生,抑制肾小管的氧化应激,但本实验中并未描述DIM如何调节NOX4的表达[34]。
干预NOX4上游信号有研究发现[24],MG149可有效抑制MYST1,而MYST1是组蛋白H4K16乙酰转移酶,与MRTF-A构成复合体,调节巨噬细胞基因的表达。使用MG149抑制MYST1,可通过MRTF-A-Myst1-NOX通路发挥对AKI的保护作用。
恩格列净通过作用于β羟基丁酸盐而抑制NOX的表达,但是由于相关的炎症因子并未减少,因此并不能减轻炎症[35],提示恩格列净对AKI的保护作用可能并不依赖炎症通路。
小结:NOX4是AKI的发病机制中的重要环节,了解NOX4在AKI中的作用对于开发AKI的新型治疗药物有重要的意义,以NOX4为靶点的治疗策略也引起了越来越多的关注。目前研究提示NOX4通过不同信号通路在SAKI、顺铂相关AKI、IRI相关AKI等疾病中发挥了关键作用,但也有研究认为NOX4是AKI的保护性因素。今后有赖于更多研究对不同AKI类型、不同病程阶段下NOX4和ROS表达的动态变化加以观察,以寻找恰当的治疗时机和干预强度,并在临床试验中进一步验证,为潜在的临床转化过程奠定基础。
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10
昆明医科大学学报(2022年4期)2022-05-23
上海交通大学学报(医学版)(2022年3期)2022-05-05
科学与财富(2021年33期)2021-05-10
作文成功之路·小学版(2020年6期)2020-07-27
世界科学技术-中医药现代化(2020年2期)2020-07-25
作文成功之路·小学版(2020年5期)2020-06-11
健康之家(2020年15期)2020-05-08
中国医学创新(2019年9期)2019-08-19
滨州医学院学报(2016年2期)2016-05-27
