
1-脱氧野尻霉素的N-取代苯丙烷衍生物的合成
2022-04-27 10:06王峰峰方志杰
化学与生物工程 2022年4期
王峰峰,方志杰
(南京理工大学化学与化工学院,江苏 南京 210014)
1-脱氧野尻霉素(DNJ)的化学名为(2R,3R,4R,5S)-2-羟甲基哌啶-3,4,5-三醇,在一系列治疗糖尿病的药物中,DNJ是国际上公认的副作用较小的生物制剂之一。DNJ及其衍生物可作为治疗糖尿病、病毒感染、肿瘤转移及溶酶体储存紊乱等疾病的药物。近年来,研究人员采用多种方法对DNJ结构进行修饰,最主要的手段是在DNJ中引入不同的疏水性基团来增强其亲脂性[1],开发了一系列结构各异的DNJ衍生物,其中N-取代衍生物一直是研究热点之一。目前已设计了一系列具有不同生物活性的DNJ的N-取代衍生物[2-4],一部分DNJ衍生物已经商品化,但是绝大多数仍处于临床研究前期或产品开发阶段。DNJ的N-取代苯丙基衍生物不仅可治疗多种病毒(如艾滋病毒、登革热病毒等)感染,还能显著抑制癌细胞转移。目前报道的DNJ的N-取代苯丙基衍生物的合成路线[5-8]都是以DNJ为起始原料,与卤代物在碱性条件下取代或与含醛基的化学试剂进行还原胺化反应,存在原料价格昂贵、含苯丙基的取代试剂合成方法繁琐、环境污染较大等缺点。鉴于此,作者以(2R,3R,4R,5S)-3,4,5-三(苄氧基)-2-((苄氧基)甲基)哌啶盐酸盐(Ⅰ)为起始原料,与取代苯乙酮(Ⅱa~Ⅱc)及多聚甲醛在无水乙醇中经曼尼希反应缩合得到中间体1-(4-取代苯基)-3-((2R,3R,4R,5S)-3,4,5-三(苄氧基)-2-((苄氧基)甲基)哌啶-1-基)丙烷-1-酮(Ⅲa~Ⅲc),再经Pd(OH)2/C加氢、脱苄、脱氧得到DNJ的N-取代苯丙烷衍生物(Ⅳa~Ⅳc),通过1HNMR、13CNMR、LC-MS对产物结构进行表征。合成路线如图1所示。
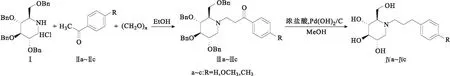
图1 DNJ的N-取代苯丙烷衍生物的合成路线
1 实验
1.1 试剂与仪器
苯乙酮、多聚甲醛、无水乙醇、甲醇、浓盐酸,分析纯,国药集团化学试剂有限公司;4-甲氧基苯乙酮、4-甲基苯乙酮,分析纯,上海麦克林生化科技有限公司;20%Pd(OH)2/C,分析纯,陕西瑞科新材料股份有限公司。
SHZ-D(Ⅲ)型循环水式多用真空泵、RE-2000型旋转蒸发器、98-2型磁力搅拌器,上海积坤实验仪器有限公司;JA2003型电子天平,上海舜宇恒平科学仪器有限公司;DFY-5L/25型低温恒温浴、DZF-6050型真空干燥箱,卯氏(上海)实验仪器有限公司;HWCL-3型集热式恒温磁力搅拌浴,郑州长城科工贸有限公司。
1.2 方法
1.2.1 1-(4-取代苯基)-3-((2R,3R,4R,5S)-3,4,5-三(苄氧基)-2-((苄氧基)甲基)哌啶-1-基)丙烷-1-酮(Ⅲa~Ⅲc)的合成
将30 mL无水乙醇、3.0 g(5.36 mmol)(2R,3R,4R,5S)-3,4,5-三(苄氧基)-2-((苄氧基)甲基)哌啶盐酸盐(Ⅰ)、8.32 mmol取代苯乙酮(Ⅱa~Ⅱc)、0.25 g(8.32 mmol)多聚甲醛加入50 mL反应罐中,85 ℃油浴反应8 h后,降至室温,加入1 mL氨水调节pH值至8~9,减压旋干,用200~300目硅胶拌样过柱,洗脱剂为石油醚(PE)-乙酸乙酯(EA) (10∶1,体积比,下同),洗脱液减压旋干,得到化合物Ⅲa~Ⅲc。
1.2.2 DNJ的N-取代苯丙烷衍生物(Ⅳa~Ⅳc)的合成
将1.0 g化合物Ⅲa~Ⅲc、40 mL甲醇加入反应瓶中,搅拌溶解,加入0.6 mL浓盐酸、150 mg 20%Pd(OH)2/C,依次用氮气置换6次、氢气置换6次,20~30 ℃保温反应50 h,过滤,滤饼分别用5 mL甲醇、5 mL水淋洗,加入20%碳酸钠溶液调节pH值至8~9,减压旋干,加入5 mL甲醇充分搅拌后,过滤除去不溶物,滤液减压旋干,用200~300目硅胶拌样过柱,洗脱剂为EA-甲醇(5∶1),洗脱液减压旋干,得到化合物Ⅳa~Ⅳc。
2 结果与讨论
2.1 合成工艺优化
2.1.1 投料量对收率的影响
以化合物Ⅲa的合成为例,在固定化合物Ⅰ投料量为1.0 eq.的情况下,考察苯乙酮与多聚甲醛的投料量对化合物Ⅲa收率的影响,结果如表1所示。
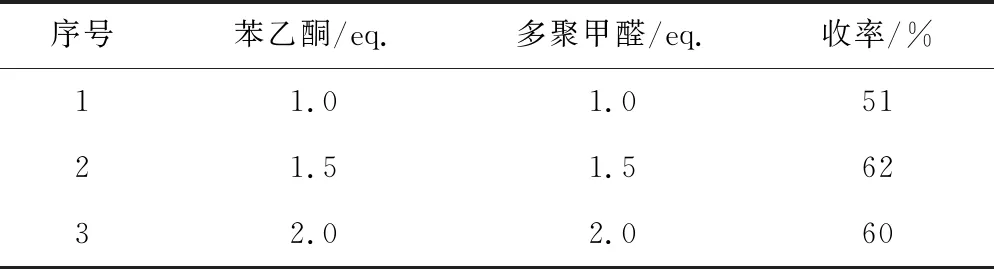
表1 苯乙酮与多聚甲醛的投料量对化合物Ⅲa收率的影响
由表1可知,当苯乙酮与多聚甲醛的投料量均为1.0 eq.时,化合物Ⅲa收率为51%;当二者的投料量均增至1.5 eq.时,收率提高到62%;继续增加二者的投料量至2.0 eq.时,收率反而有所下降,为60%,可能是由于生成了较多的副产物。因此,选择苯乙酮与多聚甲醛的投料量均为1.5 eq.。
2.1.2 反应时间对收率的影响
在化合物Ⅰ与苯乙酮、多聚甲醛的物质的量比为1∶1.5∶1.5、反应温度为80~85 ℃的条件下,考察反应时间对化合物Ⅲa收率的影响,结果如表2所示。

表2 反应时间对化合物Ⅲa收率的影响
由表2可知,当反应时间为2 h时,化合物Ⅲa收率为15%;随着反应时间的延长,收率逐渐升高,当反应时间为8 h时,收率达到最高,为62%;继续延长反应时间至16 h,收率变化不明显。因此,选择反应时间为8 h。
2.2 结构表征
1-苯基-3-((2R,3R,4R,5S)-3,4,5-三(苄氧基)-2-((苄氧基)甲基)哌啶-1-基)丙烷-1-酮(Ⅲa):白色固体,收率62.7%。1HNMR(DMSO,400 MHz),δ:7.97(d,J=7.2 Hz,2H),7.64(t,J=7.4 Hz,1H),7.52(t,J=7.7 Hz,2H),7.36~7.22(m,18H),7.19~7.15(m,2H),4.90~4.84 (m,1H),4.80~4.65 (m,3H),4.62~4.56(m,1H),4.47~4.33(m,3H),3.68(d,J=9.9 Hz,1H),3.58(dd,J=10.5 Hz、3.8 Hz,1H),3.50~3.36(m,2H),3.31~3.26(m,1H),3.24~3.10(m,4H),3.06~2.93(m,1H),2.45(dd,J=9.3 Hz、1.9 Hz,1H),2.23(t,J=10.5 Hz,1H);LC-MS,m/z:656[M+H]+。
1-(4-甲氧基苯基)-3-((2R,3R,4R,5S)-3,4,5-三(苄氧基)-2-((苄氧基)甲基)哌啶-1-基)丙烷-1-酮(Ⅲb):白色固体,收率56.5%。1HNMR(DMSO,400 MHz),δ:7.95~7.89(m,2H),7.40~7.23(m,18H),7.14(d,J=7.7 Hz,2H),6.96~6.90(m,2H),5.02~4.96(m,1H),4.93~4.81(m,2H),4.74~4.63(m,2H),4.57~4.50(m,1H),4.49~4.39(m,2H),3.90(s,3H),3.74~3.66(m,3H),3.58(t,J=9.2 Hz,1H),3.51(t,J=9.0 Hz,1H),3.30~3.22(m,1H),3.18~3.11(m,2H),3.08~2.99(m,2H),2.44(d,J=9.3 Hz,1H),2.33(t,J=10.8 Hz,1H)。
1-(4-甲基苯基)-3-((2R,3R,4R,5S)-3,4,5-三(苄氧基)-2-((苄氧基)甲基)哌啶-1-基)丙烷-1-酮(Ⅲc):白色固体,收率57.4%。1HNMR(DMSO,400 MHz),δ:7.90~7.83(m,2H),7.37~7.20(m,18H),7.08~6.95(m,2H),6.90~6.84(m,2H),4.82~4.73(m,1H),4.69~4.62(m,2H),4.59~4.51(m,2H),4.45~4.30(m,3H),3.69~3.60(m,3H),3.50(s,1H),3.42(t,J=9.0 Hz,1H),3.26~3.20(m,1H),3.15~3.03(m,2H),2.97~2.83(m,2H),2.42(d,J=9.0 Hz,1H),2.38(s,3H),2.12(t,J=10.5 Hz,1H)。
(2R,3R,4R,5S)-2-(羟甲基)-1-(3-苯基丙基)哌啶-3,4,5-三醇(Ⅳa):黄色固体,收率74.6%。1HNMR(DMSO,400 MHz),δ:7.31~7.14(m,5H),4.70(s,3H),4.15(s,1H),3.71(d,J=11.2 Hz,1H),3.54(d,J=10.6 Hz,1H),3.25~3.17(m,1H),3.05(t,J=8.8 Hz,1H),2.92(t,J=8.8 Hz,1H),2.85~2.74(m,2H),2.60~2.41(m,3H),2.04~1.95(m,2H),1.75~1.62(m,2H);LC-MS,m/z:282 [M+H]+。
(2R,3R,4R,5S)-2-(羟甲基)-1-(3-(4-甲氧基苯基)丙基)哌啶-3,4,5-三醇(Ⅳb):黄色固体,收率65.2%。1HNMR(DMSO,400 MHz),δ:7.15~7.07(m,2H),6.85~6.81(m,2H),4.75~4.62(m,3H),4.13(s,1H),3.72(s,3H),3.68(s,1H),3.58~3.50(m,1H),3.26~3.17(m,1H),3.05(td,J=8.9 Hz、4.7 Hz,1H),2.92(t,J=8.3 Hz,1H),2.84~2.72(m,2H),2.50~2.37(m,3H),2.02~1.91(m,2H),1.71~1.59(m,2H);13CNMR(DMSO,101 MHz),δ:157.77,134.52,129.56,114.11,79.62,71.24,69.88,67.14,59.50,57.39,55.42,52.16,32.63,27.14。
(2R,3R,4R,5S)-2-(羟甲基)-1-(3-(4-甲基苯基)丙基)哌啶-3,4,5-三醇(Ⅳc):黄色固体,收率70.0%。1HNMR(DMSO,400 MHz),δ:7.07(s,4H),4.74~4.59(m,3H),4.11(s,1H),3.70(d,J=11.3 Hz,1H),3.58~3.49(m,1H),3.26~3.17(m,1H),3.10~3.00(m,1H),2.91(td,J=8.7 Hz、3.7 Hz,1H),2.84~2.71(m,2H),2.50~2.36(m,3H),2.26(s,3H),2.01~1.88(m,2H),1.73~1.60(m,2H);13CNMR(DMSO,101 MHz),δ:139.56,134.86,129.26,128.53,79.65,71.28,69.91,67.16,59.57,57.42,52.20,33.12,27.02,21.09。
2.3 反应机理
基于实验结果和已有的文献报道,其可能的反应机理(图2)为:在酸性条件下羰基发生质子化,胺基上的孤对电子对羰基进行亲核加成,氮上的质子转移到羟基上,在脱水的同时氮上的电子发生转移,得到一个亚胺离子中间体,亚胺离子进攻含活泼氢化合物的烯醇型结构,同时失去质子,最终发生氨甲基化反应,得到中间体Ⅲ;中间体Ⅲ经催化加氢,羰基被还原为羟基,在酸性条件下,苄位羟基质子化脱水形成碳正离子,再进一步夺取氢负离子同时脱去质子,得到目标化合物Ⅳ。
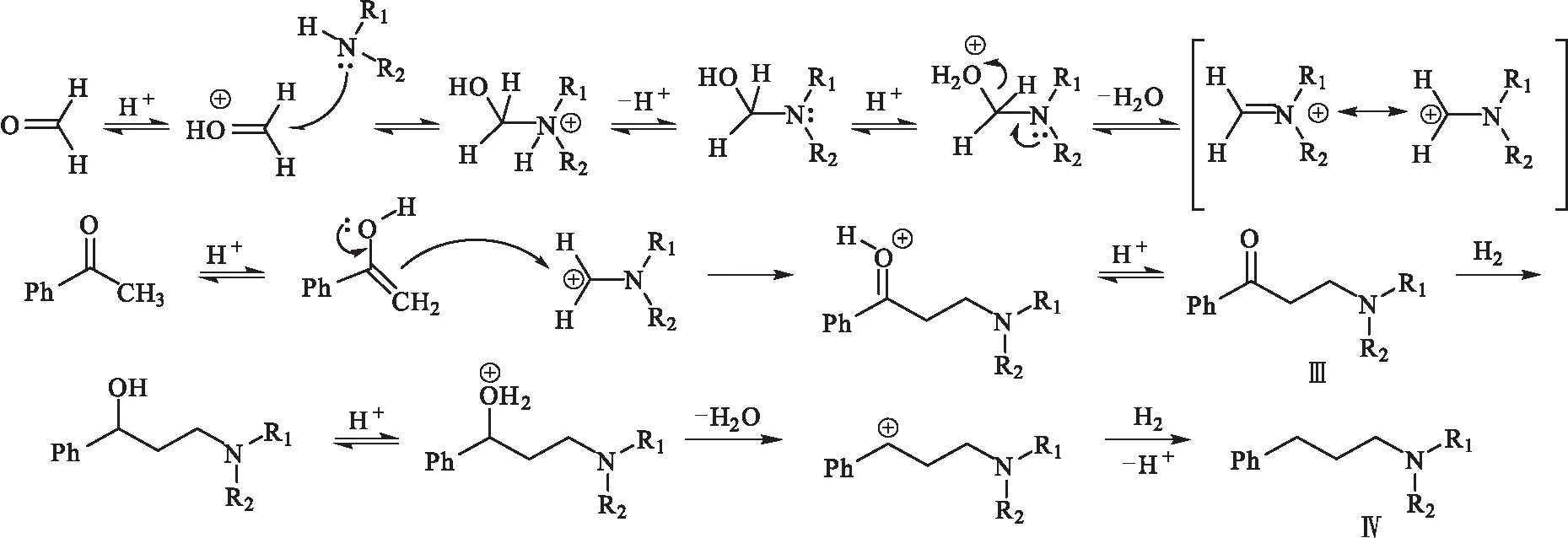
图2 目标化合物可能的反应机理
3 结论
以(2R,3R,4R,5S)-3,4,5-三(苄氧基)-2-((苄氧基)甲基)哌啶盐酸盐为起始原料,经曼尼希缩合、催化氢化、脱苄、脱氧得到DNJ的N-取代苯丙烷衍生物。该方法原料易得、反应步骤简单、环境污染小,后续可将苯环替换为其它芳香环合成多种类型的DNJ的N-取代衍生物。
猜你喜欢
工业催化(2022年9期)2022-10-17
化工生产与技术(2022年3期)2022-07-01
健康体检与管理(2022年2期)2022-04-15
化学工业与工程(2022年1期)2022-03-29
浙江大学学报(理学版)(2021年5期)2021-09-17
食品安全导刊(2021年20期)2021-08-30
精神医学杂志(2021年2期)2021-06-23
安徽化工(2021年3期)2021-05-29
能源工程(2021年1期)2021-04-13
安徽化工(2018年3期)2018-07-04
