
STAT3抑制剂抗肿瘤研究进展
2022-04-15 03:03黎勇玲孙振亮
药学进展 2022年3期
黎勇玲 ,孙振亮
(1. 南方医科大学药学院,广东 广州 510515;2. 南方医科大学附属奉贤医院国家药物临床试验中心,上海 201499)
信号转导和转录激活因子(STAT)为细胞质转录因子,可将信号从细胞中的生长因子和细胞因子传递到细胞核。STAT家族包含7个成员:STAT1~STAT4、STAT6,以及STAT5的2个亚型,即STAT5a和STAT5b。研究证实,STAT3与STAT家族的其他成员都有一个特殊的三维结构,其特征是含有6个主要结构域:1)氨基末端结构域(NTD);2)卷曲螺旋结构域(CCD);3)DNA结合结构域(DBD);4)连接域(LD);5)Src同源2(SH2)结构域;6)反式激活结构域(TAD)(见图1)[1]。
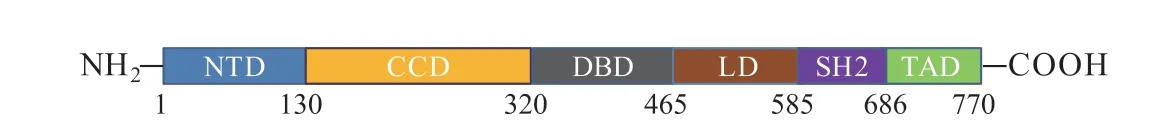
图 1 STAT3域示意图Figure 1 Schematic diagram of STAT3 domain
STAT3由770个氨基酸组成[2]。每个结构域在信号转导和基因转录活化的过程中发挥着不同的作用。NTD可以促进蛋白质与蛋白质的相互作用。CCD具有核定位作用,DBD是STAT3蛋白和DNA相互作用所必需的场所。LD 参与连接DNA的结构域与SH2结构域,保证DNA结构域的稳定性。SH2结构域识别靶蛋白中的酪氨酸磷酸化,是STAT3二聚化所必需的场所,而TAD包含1个丝氨酸,它可以通过磷酸化来促进其他转录激活因子的组装[3]。此外,NTD还被发现对癌细胞中促凋亡基因的转录具有抑制作用[4]。
在未受刺激的细胞中,STAT3受到负调节因子如活化STAT蛋白抑制剂(PIAS)、细胞因子信号转导抑制因子(SOCS)家族、蛋白酪氨酸磷酸酶和泛素酶的严格调控,以维持在细胞质中的非活性状态[5]。STAT3主要通过其上游配体诱导的酪氨酸残基的直接磷酸化而激活,这些配体包括Janus激酶(JAK)、酪氨酸激酶、细胞因子和非受体酪氨酸激酶。活化的STAT3单体通过SH2形成二聚体结构,并通过核转运蛋白转移至细胞核,与核内特定DNA序列结合,行使基因转录调控的功能。除了磷酸化,其他翻译后修饰(即乙酰化、甲基化和泛素化)也可以通过改变STAT3磷酸化来调节STAT3的转录活性[6]。STAT3发挥细胞功能的机制被总结为3个步骤:1)特定激酶的蛋白磷酸化;2)磷酸化促进STAT3的二聚化;3)磷酸化的STAT3二聚体激活基因表达。
STAT3可在多种人类肿瘤中被激活,包括实体肿瘤和血液学肿瘤,在多种患者来源的肿瘤组织样本中观察到STAT3过表达,其可能导致疾病的不良预后。相比之下,使用抑制剂或敲除系统阻断STAT3激活可以显著抑制肿瘤进展,从而强调阻断STAT3信号级联在癌症治疗中的重要性[7]。本文主要综述近几年STAT3抑制剂的抗肿瘤研究进展。
1 STAT3上游信号抑制剂
抑制STAT3激活的一个有效策略是抑制上游组分,主要集中在抑制JAK、Src和受体酪氨酸激酶(RTK)方面。STAT3的间接抑制剂与这些组分相互作用,抑制STAT3的激活,进而导致STAT3调控基因表达失败。
1.1 Janus激酶抑制剂
JAK是STAT3最重要的上游信号因子,在某些应激反应条件下,一些细胞因子与相应的受体结合,诱导JAK磷酸化,从而使STAT3活化,促使STAT3的二聚化、核易位、与特定的DNA序列结合,行使基因转录调控的功能(见图2)。因此,JAK抑制剂是最主要的间接性STAT3抑制剂,通过抑制STAT3活化而发挥作用。鲁索利替尼(ruxolitinib)、托法替尼(tofacitinib)和帕利替尼(pacritinib)是目前研究最广泛的JAK抑制剂。到目前为止,JAK抑制剂的临床应用主要集中在涉及慢性炎症和骨髓增生性肿瘤等疾病方面,对用于实体瘤患者的评估较少[8]。鲁索利替尼是首个被批准的选择性JAK1/2抑制剂,用于治疗骨髓纤维化和真性红细胞增多症[9]。托法替尼是选择性JAK1/3抑制剂,用于治疗类风湿性关节炎,目前正在接受治疗溃疡性结肠炎、克罗恩病和牛皮癣的临床评估[10-12]。
AZD1480是一种小分子JAK1/2抑制剂,其可通过阻断STAT3信号有效地抑制实体肿瘤的发生。然而,对实体肿瘤患者的Ⅰ期临床评估显示,使用AZD1480治疗后发生了神经不良反应,包括焦虑、共济失调、行为变化、幻觉和记忆丧失,导致该药物的进一步评估中止[13]。
1.2 Src抑制剂
Src是一种与细胞膜相关的非受体酪氨酸激酶,在肿瘤细胞的增殖、迁移和分化过程中发挥关键作用。在分子水平上,Src诱导酪氨酸残基磷酸化从而使STAT3活化,促进STAT3二聚化并通过核转运蛋白转移至细胞核,与核内特定DNA序列结合,行使基因转录调控的功能(见图2)。因此,Src也被认为是癌症治疗的一个有吸引力的治疗靶点。Bcr-abl酪氨酸激酶融合蛋白是由9号和22号染色体相互易位产生的基因编码的一种Src激酶,已证明Bcr-abl融合基因是诱导造血干细胞转化为慢性粒细胞白血病(CML)所必需的[14]。甲磺酸伊马替尼是第一个Bcr-abl抑制剂,它于2001年被批准用于治疗CML,到目前为止一直是这些患者的标准治疗方法。尽管伊马替尼有积极作用,但临床试验数据显示,伊马替尼既能引发耐药性,又有一定毒性而导致耐受不良。为了解决这些问题,达沙替尼、尼洛替尼和波苏替尼作为Bcr-abl的第二代抑制剂被开发出来,并获FDA批准用于治疗对伊马替尼耐药的CML[8]。口服Src抑制剂达沙替尼在复发性上皮卵巢癌患者中单独使用的疗效有限[15]。然而,达沙替尼与紫杉醇联合使用可协同抑制卵巢肿瘤细胞的生长[16]。
1.3 受体酪氨酸激酶抑制剂
RTK可与一些受体结合,使自身发生磷酸化,从而诱导STAT3激活(见图2)。RTK的受体包括表皮生长因子受体(EGFR)、成纤维细胞生长因子受体(FGFR)、人类表皮生长因子受体2(HER2)、肝细胞生长因子受体(HGFR)、血小板衍生生长因子受体(PDGFR)、血管生长因子受体(VEGFR)和胰岛素样生长因子受体(IGFR)等[17]。卡博替尼(cabozantinib)是一种VEGFR抑制剂,Xiang等[18]研究表明cabozantinib可以削弱STAT3的激活和功能,从而抑制肿瘤生长。VEGFR抑制剂阿昔替尼(axitinib)已被证明可减少树突状细胞中STAT3的磷酸化,并损害其表型和功能[19]。阿帕替尼(apatinib)是一种具有高度选择性的VEGFR2抑制剂,已被证明可通过靶向STAT3抑制骨肉瘤的迁移和侵袭以及程序性死亡蛋白配体1(PD-L1)的表达[20]。

图 2 STAT3 激活机制示意图Figure 2 Diagram of STAT3 activation machanism
2 STAT3直接抑制剂
直接靶向STAT3的最常见的方法是通过破坏SH2、DBD或NTD的结构域来防止功能性STAT3二聚体的形成。一般说来,STAT3的直接抑制剂可以分为3类:肽类、寡核苷酸类和小分子类。
2.1 肽类
多肽类药物通常是根据STAT3蛋白中氨基酸残基的结构设计的,可以针对不同的结构域。磷酸肽抑制剂PY*LKTK来源于STAT3 SH2结构域的结合肽序列,是首次成功地破坏STAT3二聚化的尝试[21]。然而,由于肽类药物细胞通透性差,体内不稳定,目前其临床应用的进一步开发受到限制。
类似地,ISS-610、PM-73G和S3I-1757等肽类化合物也均来源于STAT3 SH2结构域结合肽序列,与STAT3的SH2结构域结合,削弱STAT3的激活和二聚化[22-23]。这些基于肽的药物抑制细胞生长并诱导肿瘤细胞凋亡,但由于它们缺乏稳定性、膜渗透性,以及有潜在的免疫原性,效力有限,还未达到进入临床试验的条件[24]。
2.2 寡核苷酸类
寡核苷酸是针对STAT3靶标的一种新的治疗策略。与STAT3结合的诱饵寡核苷酸可以隔离STAT3,从而减少其与靶基因内同源DNA位点的结合[25]。反义寡核苷酸(ASO)旨在通过靶向STAT3 mRNA来阻断STAT3的活性。例如,第2代STAT3反义寡核苷酸AZD9150针对STAT3基因的3'-非翻译区(3'-UTR)[26]。临床前试验和临床评估表明,AZD9150在肿瘤治疗中具有高效、低毒的特点。尽管STAT3的寡脱氧核苷酸抑制剂具有很好的特异性和效价,但其细胞膜透过性差、降解速度快、缺乏有效的靶向递送载体,仍然是阻碍其在实体瘤中应用的主要障碍。而适体因其体积小、稳定性高和免疫原性低等优点成为常规药物和小RNA(包括siRNA和miRNA)的有效靶向递送剂。研究发现适体siRNA嵌合体介导的STAT3沉默在胶质母细胞瘤的治疗中具有很好的抑制作用,提示改良的寡核苷酸可能具有良好的实体瘤治疗前景[27]。
2.3 小分子类
小分子STAT3抑制剂可与STAT3的不同结构域相互作用,抑制STAT3的激活和功能。口服小分子化合物HJC0152通过抑制Tyr705残基的磷酸化使STAT3失活。体外实验表明,HJC0152能抑制胶质母细胞瘤细胞的增殖和迁移,诱导细胞凋亡,且增强胶质母细胞瘤细胞的化疗敏感性[28]。
Wang等[29]研究发现,一种基于隐丹参酮改造的STAT3抑制剂LYW-6下调了下游癌基因的表达,从而使大肠癌细胞周期阻滞于G1期,并有效地增加了细胞凋亡,还阻断了侵袭和转移相关的信号转导通路。此外,研究人员在异种移植模型中,观察到LYW-6显著降低肿瘤组织中STAT3的磷酸化水平,并显著抑制肿瘤生长。在化学诱导的结直肠癌模型中,LYW-6治疗也能明显抑制肿瘤的发展。
Huang等[30]通过虚拟筛选发现了一种针对STAT3 DNA结合域的新型小分子STAT3抑制剂InS3-54。InS3-54可以特异性地抑制肿瘤细胞的增殖、侵袭和转移。此外,通过进一步的研究,课题组还发现了一种改进的先导化合物InS3-54A18具有更高的特异性和药理活性。InS3-54A18不仅在体外和体内直接与DBD结合并抑制STAT3的DNA结合活性,而且还可有效抑制STAT3下游靶基因的表达,以及抑制肺移植瘤的生长[31]。
BBI608(napabucasin)是一种可选择性地与STAT3的DBD结构域结合的小分子抑制剂,是迄今为止唯一进入Ⅲ期试验的直接STAT3抑制剂[32]。最近的一项BBI608的单药治疗Ⅲ期临床试验结果表明,BBI608在晚期结直肠癌治疗中有潜在的应用价值。
通过虚拟筛选,研究人员发现了许多STAT3的小分子抑制剂,尽管这些抑制剂在体外表现出很好的物理化学性质,但它们中的大多数临床疗效并不理想,这可能是因为其水溶性和细胞通透性较低。最近,基于小分子蛋白水解靶向嵌合体(PROTAC)的策略已经引起了人们的广泛关注,因为它既可以抑制靶蛋白的功能,又可以阻断靶蛋白的增加。王少萌教授团队研制的小分子SD-36,其为STAT3的选择性降解剂。SD-36含有2个官能团:STAT3 SH2结构域的小分子结合体(SI-109)和Cereblon(CRBN)E3连接酶的配体(来那度胺)。当SI-109与STAT3的SH2结构域结合后,CRBN被招募,而CRBN又招募E2酶来促进STAT3的泛素化。泛素化的STAT3则以蛋白酶体降解为目标。SD-36在细胞系和异种移植小鼠模型中都显示出良好的效果[33-34]。
3 结语
目前在研的STAT3抑制剂如表1所示。

表1 在研STAT3抑制剂一览Table 1 The inhibitors of STAT3 in development
STAT3是重要的转录因子,参与多种生物学功能,包括细胞增殖、存活、凋亡和炎症。而STAT3异常激活可诱导肿瘤的发生和发展。以STAT3为靶点,通过阻断上游信号通路间接抑制STAT3,或通过肽、寡核苷酸和小分子直接抑制STAT3的策略,均是通过干扰STAT3的磷酸化来阻止功能性STAT3二聚体的形成来影响肿瘤的发生发展。
到目前为止,许多STAT3抑制剂在体内外实验中取得了十分显著的效果,且部分抑制剂已经开展临床试验。然而,大多数小分子抑制剂由于水溶性和细胞通透性较低而表现出较差的临床疗效。在未来的研究中,PROTAC可作为开发有效STAT3抑制剂的一个新思路——其既可以抑制靶蛋白的功能,又可以阻断靶蛋白表达,这种双重功能使其具有强的药效和更高的选择性,可以克服一般STAT3抑制剂临床疗效有限、且存在一定毒副作用的问题。研究人员需对STAT3蛋白进行更深入研究,并进一步探索PROTAC对不同细胞环境和各种类型肿瘤的作用。
猜你喜欢
中风与神经疾病杂志(2022年9期)2022-10-19
保健与生活(2022年5期)2022-03-15
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
科学24小时(2018年1期)2018-01-10
分析化学(2017年12期)2017-12-25
现代养生·下半月(2016年6期)2016-10-21
恋爱婚姻家庭·青春(2016年10期)2016-10-10
中国美容医学(2004年3期)2004-09-17
