
ANGPTL3与动脉粥样硬化
2022-04-12 00:35张月李玥王丽洪斌
中国医药生物技术 2022年2期
张月,李玥,王丽,洪斌
心血管疾病(cardiovascular disease,CVDs)是全世界普遍存在的一种疾病,其发病率和死亡率均居于首位[1]。高脂血症作为发生此类疾病的高危因素之一,可直接引起严重的心血管疾病,例如动脉粥样硬化等。其中脂蛋白代谢紊乱,血浆脂质如低密度脂蛋白胆固醇(low-density lipoprotein cholesterol,LDL-C)异常升高可增加动脉内斑块的聚集,提高动脉粥样硬化性心血管疾病的患病风险。有研究显示,血浆甘油三酯(triglycerides,TG)的升高也是导致动脉粥样硬化发生的风险因子[2-3]。随着人们生活方式的改变和生活水平的提高,心血管疾病逐渐年轻化,因此,早期发现和治疗对于降低心血管疾病的发生和发展至关重要[1]。近几年已经有多个有效降低 LDL-C 的药物上市,包括他汀类药物和 PCSK9 抑制剂,然而针对降低血浆 TG 的疗法并不多。血管生成素样蛋白 3(angiopoietin-like protein 3,ANGPTL3)自被发现以来,一直受到人们的关注。已有文献报道,ANGPTL3 可通过抑制脂蛋白脂肪酶(lipoprotein lipase,LPL)和内皮脂肪酶(endothelial lipase,EL)等的活性,导致血浆 TG 及 LDL-C 水平升高[4-6]。人类遗传学研究表明,angptl3 基因天然缺失的杂合子个体的心血管疾病患病风险下降约 40%,且其血浆 LDL-C 和 TG 水平都有显著下降。因此,ANGPTL3 被认为在脂蛋白代谢调控中发挥了关键的作用,成为调节血浆脂质水平和降低心血管疾病风险的重要研究靶标[7]。本文将从以下几个方面进行综述。
1 ANGPTL3 概述
1.1 ANGPTL3 的发现
ANGPTL3 是 1999年由 Conklin 等[8]首次提出,其属于血管紧张素样蛋白家族中的一员,该蛋白家族因其结构与血管紧张素相似而得名。ANGPTL 基因家族编码一类分泌性蛋白,即 ANGPTL1 ~ 8,它们具有相似的结构且一系列研究证实部分成员在血脂代谢中发挥重要作用(表1)[9-10]。2002年,Koishi 等[11]首次发现了糖尿病模型 KK 小鼠的一种突变小鼠 KK/San,该品种小鼠angptl3 的 6 号外显子中插入了 4 bp 的序列,使 ANGPTL3 蛋白翻译提前终止而失活,从而产生低脂血症的表型,重新引入完整的angptl3cDNA 后得以缓解;进一步研究发现 ANGPTL3 可通过抑制 LPL 活性来调节 TG 水平[4];2009年,Romeo等[12]发现 ANGPTL3 的突变可通过干扰蛋白质的合成与分泌或失去抑制 LPL 活性的能力,显著影响人的血浆甘油三酯水平;随后哈佛医学院等单位联合发现 2 例 ANGPTL3功能缺失突变体,他们健康状况良好且伴有家族性合并低脂血症(低 LDL-C、HDL-C 和甘油三酯)[13];2017年 4月,JACC报道 ANGPTL3 功能失活突变体可显著降低心血管疾病发病率[14];上述研究结果证实 ANGPTL3 在脂质代谢中的关键作用且提示 ANGPTL3 是一个安全的降血脂药物靶点。

表1 ANGPTLs 的血脂代谢功能
1.2 ANGPTL3 蛋白的结构
ANGPTL3、ANGPTL4 及 ANGPTL8 均与血脂代谢相关且具有相似的结构(图1)。ANGPTL3 的结构在不同种属中有些许差别,人类 ANGPTL3 是含有 460 个氨基酸的多肽,基因定位于 1 号染色体 1p31 区域;小鼠 ANGPTL3是由 4 号染色体上 7 个外显子编码的含有 455 个氨基酸的多肽,全长约 11 kb,与人 ANGPTL3 多肽的同源性为76%。ANGPTL3 结构中包括 N 端信号肽序列、N 端螺旋结构域、连接区和 C 端纤维蛋白原样结构域(图1)[15-17]。N 端螺旋结构域可通过增强前蛋白转化酶切割 LPL 的活性,裂解 LPL 从而抑制 LPL 活性,增加血浆 TG 水平[18]。C 端纤维蛋白原样结构域可与整合蛋白 αvβ3 受体结合,发挥血管生长素作用。连接区域中存在弗林蛋白酶(furin)的两个切割位点,即 Arg221—Ala222和 Arg224—Thr225[15]。
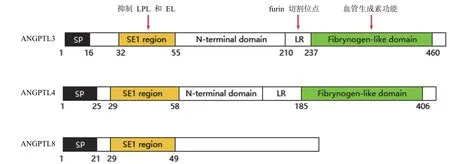
图1 ANGPTL3/4/8 的结构示意图[15-17]
1.3 angptl3 基因的转录调控
ANGPTL3 是主要在肝脏表达的一种分泌蛋白,在肝脏发育早期即有表达,且于成人肝脏中维持表达;ANGPTL3在肾脏足细胞中也有少量表达,但其功能尚不清楚[8]。在转录水平,angptl3 主要受肝脏 X 受体(liver X receptors,LXRs)和肝细胞核因子 1α(hepatocyte nuclear factor 1α,HNF1α)等调控[19-22]。其中,LXRs 主要包含两种受体,LXRα和 LXRβ。LXRα 因其最初是在人类肝脏 cDNA 文库中分离而得名,主要在肝脏和其他参与脂质代谢的组织中表达,LXRβ 几乎在所有组织中表达[23]。早期 LXRs 已报道最主要的作用是通过上调 CYP7A1 表达以增加胆汁酸/胆固醇输出,对维持体内胆固醇平衡发挥重要调节作用[23]。研究发现,在angptl3启动子区存在 LXRs 反应元件(LXR response elements,LXREs),且高胆固醇饲料可通过激活LXREs 增加小鼠肝脏中angptl3 表达,从而使血浆甘油三酯水平也相应升高[19]。而在angptl3缺陷的 C57BL/6J 突变小鼠中,却没有观察到上述现象[20]。说明 LXRs 在angptl3 表达过程中发挥重要调控作用。
此外,有研究团队发现,甲状腺激素以甲状腺激素受体 β(thyroid hormone receptor β,TRβ)依赖的方式抑制angptl3 表达。后续研究表明,angptl3 启动子的近端(-171 ~ +66)区域中转录因子 HNF1 结合位点在 TRβ 诱导的angptl3抑制中发挥了关键的作用,TRβ 通过抑制HNF1α 的转录活性进而拮抗其介导的信号通路[22]。除甲状腺激素外,胰岛素与瘦素也是参与angptl3表达调控的负调控因子[21],但其参与调控的转录因子及调控机制知之甚少。
1.4 ANGPTL3 蛋白的翻译后修饰
ANGPTL3 蛋白具有多种翻译后修饰方式,如ANGPTL3 蛋白翻译后剪切以及 O-糖基化修饰作用,通过不同的翻译后水平调控从而影响 ANGPTL3 蛋白的表达或稳定性。ANGPTL3 翻译后剪切是由 Ono 等[15]首次提出,他们发现 ANGPTL3 蛋白连接区的 Arg221—Ala222和Arg224—Thr225两个位点可被前蛋白转化酶剪切,且经剪切后的 ANGPTL3 的 N 末端螺旋结构域抑制 LPL 的活性更强,因此 ANGPTL3 的翻译后剪切被认为对于ANGPTL3 的活化至关重要。而在前蛋白转化酶识别位点,紧邻 C 末端处包含两个潜在的 GalNAc O-糖基化位点(TT226),GalNAc-转移酶通过对其 O-糖基化修饰阻止前蛋白转化酶对裂解位点的识别,抑制 ANGPTL3 的降解[24],因此 TT226位的糖基化可显著影响 ANGPTL3 的活性及血脂水平。
2 ANGPTL3 与动脉粥样硬化疾病发生
不同类型的人类遗传学研究已经证实 ANGPTL3 与脂质代谢的关系。利用全基因组关联研究(genome-wide association studies,GWAS)和外显子测序,证实angptl3 基因突变可以导致 ANGPTL3 蛋白功能缺失引起家族性合并低脂血症,其特征为血浆 LDL-C、HDL-C 及 TG 浓度极低[12-13,25]。目前发现angptl3 突变包括 E129X 和 S17X两种无义突变,发生突变的家庭成员血浆中 LDL-C、TG、HDL-C 均低于未突变者[13]。家族性高胆固醇血症(familial hypercholesterolemia,FH)是一种以 LDL-C 水平升高为特征的严重遗传性高脂血症,可导致过早动脉粥样硬化。研究发现,在家族性高胆固醇血症患者中罕见地出现了两种突变:外显子突变(c.A956G: p.K319R)和非翻译区突变(c.*249G > A),且在 K319R 突变患者中,与未突变患者相比,其胆固醇代谢水平及 ANGPTL3 的表达均有所上调[26]。在小鼠中观察到,ANGPTL3 表达降低可减少主动脉动脉粥样硬化斑块面积[27-28]。在探究angptl3 缺失与人类动脉粥样硬化关系时发现,angptl3 完全缺失的个体没有动脉粥样硬化发生的迹象,且突变携带者与未突变患者相比,血浆中 TG 降低 17%,LDL-C 降低 12%,冠心病发病几率降低 34%[14]。在一项人类遗传学研究中,angptl3杂合突变参与者血清中 TG、LDL-C 和 HDL-C 的浓度明显低于未突变参与者[7]。在血脂异常小鼠中,ANGPTL3 抑制剂单克隆抗体 evinacumab通过抑制 ANGPTL3 使小鼠动脉粥样硬化斑块面积和坏死区域大幅度下降[7]。在一项针对 FH 患者的临床试验中,相比于安慰剂对照组,患者在通过ANGPTL3 抑制剂 evinacumab 治疗后,可显著降低血浆LDL-C 水平[29]。这些证据有力地证实了抑制 ANGPTL3 与冠心病防治密切相关。
2.1 ANGPTL3 对脂质代谢酶的影响
目前研究认为,ANGPTL3 主要通过抑制 LPL 和 EL的活性发挥脂蛋白代谢调节作用,改变脂蛋白谱组成,使血脂水平发生变化。其中 LPL 是 TG 脂肪酶家族中的一员,位于毛细血管腔内表面,可催化 TG 水解,释放出游离脂肪酸被邻近组织吸收。TG 在脂质储存(脂肪组织)和代谢(主要是心脏和骨骼肌)之间的分配主要由 LPL 的相对活性所决定[12]。研究证明 LPL 活性改变可能与其翻译后修饰密切相关,而非转录水平的调控[18]。在细胞实验及动物实验中,ANGPTL3 均已被证实可通过增强内源性弗林和 PACE4 对 LPL 的切割作用,LPL 裂解后从细胞表面解离下来,其催化和非催化功能均被抑制,LPL 的 N端与 C 端结构域完整是 ANGPTL3 诱导其裂解的必要前提[4,18,30-31]。
除 ANGPTL3 以外,ANGPTL4 和 ANGPTL8 也被证实是 LPL 的负调节因子[32-34]。ANGPTL4 与 ANGPTL3有 31% 的同源序列(图1),但其抑制 LPL 活性的机制却不相同。ANGPTL3 主要表现出对 LPL 催化活性的可逆性抑制,而 ANGPTL4 的 N 端结构域可瞬时与 LPL结合,这种相互作用导致 LPL 从具有催化活性的二聚体转变为对肝素亲和力降低的无活性单体[30,35]。且 ANGPTL3主要在正常饮食条件下对 LPL 的抑制效率较高,而ANGPTL4 则是在禁食期间发挥较高的抑制活性[32]。ANGPTL3 与 ANGPTL8 在抑制 LPL 时发挥协同作用。ANGPTL8 的 N 端结构域与 ANGPTL3 和 ANGPTL4 具有 20% 的序列同源性,但缺乏 C 端纤维蛋白原样结构域(图1)。ANGPTL8 自身没有 LPL 抑制活性,需要与ANGPTL3 共同作用来实现。相反,在缺乏 ANGPTL8 的情况下,ANGPTL3 抑制 LPL 的活性也会受到影响。此外,在小鼠中,ANGPTL3 和 ANGPTL8 的共同表达比单独表达 ANGPTL3 可更有效地增加血浆中 TG 含量[10,36-37]。近几年,在不同饮食条件下,ANGPTL8 与ANGPTL3/ANGPTL4 形成的复合物调节不同组织内 LPL的活性及 TG 的合成代谢逐渐被关注[38-39]。
EL 是 TG 脂肪酶家族中新发现的一位成员,与其他家族成员相比,这种脂肪酶是由内皮细胞合成,因此被称为内皮脂肪酶(由 LIPG 基因编码)。EL 具有较高的磷脂酶活性,而甘油三酯脂肪酶活性较低。在体内,EL 在肝、肺、肾、胎盘等器官中均有表达,但在骨骼肌中不表达。EL 的表达部位、作用特点和体内效应表明,它可能在脂蛋白代谢和血管生物学中发挥作用[40-41]。血浆中低水平的 HDL-C 已被公认为是一种代谢综合征的表现,也是导致心血管事件的一个至关重要的危险因素[42]。目前的研究揭示了 ANGPTL3可通过抑制 EL 发挥调节人与动物的 HDL-C 及磷脂的作用[5],同时 EL 在含有 ApoB 的脂蛋白(LDL 及 VLDL)的分解代谢中也可能发挥重要作用[43]。在缺乏 LDLR 时,抑制 ANGPTL3 可以通过限制 LDL 颗粒的产生来降低LDL-C,而在机制上,EL 是该通路中的关键中介[44]。
2.2 ANGPTL3 对 LDL-C 代谢的影响
研究表明,单克隆抗体 REGN1500 可与 ANGPTL3结合并抑制后者的活性作用可以通过不影响肝脏分泌含ApoB 脂蛋白的数量而改变其颗粒组成,使其通过非经典途径从循环中更快地清除,从而降低 LDL-C 的水平[45]。然而也有研究发现,沉默angptl3 基因可通过降低 ApoB 分泌和增强 ApoB 脂蛋白摄取的双重机制降低 LDL-C 水平[46]。当 LDLR 缺失时,抑制 ANGPTL3 可驱动 VLDL重塑和清除,反过来会消耗 LDL 前体,限制 LDL 颗粒的产生,导致血浆 LDL-C 水平的降低[44]。抑制 ANGPTL3 还可以通过下调糖异生基因的表达提高胰岛素敏感性,使得富含 TG 的 VLDL-1 型颗粒向脂质稀少的 VLDL-2 型颗粒转变而影响 LDL-C 的产生[47]。除了整体降低 TG 和LDL-C 的作用,ANGPTL3 缺乏还可使富含甘油三酯的脂蛋白(triglyceride-rich lipoprotein,TRLs)及其残余物中胆固醇比例的降低[48]。
3 ANGPTL3 与降脂治疗
3.1 单克隆抗体
Evinacumab 是再生元制药公司研发的一种全人源靶向ANGPTL3 的 IgG4 亚型的的单克隆抗体,当与其他降脂疗法联合使用时,可将家族性高胆固醇血症患者中危险的高胆固醇水平降至正常水平,于 2021年 2月 11日由 FDA批准上市,用于治疗 12 岁及以上儿童或成人家族性纯合子高胆固醇血症患者。此前,FDA 授予了 evinacumab 突破性药物资格(BTD),孤儿药资格和优先审评资格,此次批准是基于 III 期临床试验研究(NCT03399786)的积极结果。
在 I 期临床试验中,与安慰剂相比,应用 evinacumab可使得 TG 呈剂量依赖性下降[49]。在针对纯合子家族性高胆固醇血症成人患者进行的一项单组、开放标签的研究中,给予 evinacumab 后,LDL-C 水平显著降低,且这些减少是在稳定、积极的降脂治疗后已经达到的基线水平之后的进一步抑制作用[50];在一项 III 期临床试验中,给药组相对于对照组,LDL-C 水平出现明显下降趋势且无不良反应事件发生[29]。在 2019年报道的两项针对高甘油三酯血症个体的 I 期试验结果显示,evinacumab 在高甘油三酯血症中耐受性良好[49]。因此,evinacumab 可以作为一种降低甘油三酯和其他脂类的治疗选择,特别是对于那些由基因突变导致高甘油三酯血症或高胆固醇血症且治疗选择有限的患者。目前,礼来公司研发靶向 ANGPTL3/8 的单克隆抗体LY3475766 正处于 I 期临床试验阶段。
3.2 RNA 疗法
反义寡核苷酸是一类序列特异地与靶基因 DNA 或mRNA 结合从而抑制该基因表达的核酸分子,早在 1978年就被提出可将其作为一种治疗疾病手段。在一项靶向angptl3 mRNA 的反义寡核苷酸(angptl3 ASO)的小鼠实验中,给药后的小鼠肝脏angptl3 mRNA、ANGPTL3 蛋白、TG 和 LDL-C 水平呈剂量依赖性下降,且伴随有肝脏 TG含量下降,动脉粥样硬化进展延缓,胰岛素敏感性增加等现象[51]。Vupanorsen 是由 Ionis pharmaceuticals 和辉瑞联合开发的一款靶向 ANGPTL3 的反义寡核苷酸疗法,用于降低肝脏中的 ANGPTL3 蛋白水平。在 I 期临床试验中,治疗组 ANGPTL3 蛋白含量、TG、LDL-C、VLDL-C、non-HDL-C 都出现了下降的趋势[28]。IIa 期临床试验中,在接受剂量为 80 mg(每四周一次)的 vupanorsen 治疗的患者与对照组相比,空腹甘油三酯水平平均下降 44%[52]。目前,IIb 期临床试验正在进行中。
siRNA 是一类长度为 20 ~ 25 个碱基对的双链 RNA,可诱发同源 mRNA 高效特异性降解,即 RNA干扰(RNA interference,RNAi)现象。目前,RNAi 疗法的相关技术也逐渐走向成熟。在 Xu 等[46]的实验中,利用 RNAi在 5 个小鼠模型和人肝癌细胞中抑制angptl3,并通过CRISPR/Cas9 基因组编辑系统删除angptl3 来验证结果。数据表明,RNAi 通过沉默angptl3 可降低小鼠体内TG、HDL-C 和 LDL-C 水平。Arrowhead 公司开发的ARO-ANG3 是一款特异性靶向肝细胞的 RNAi 疗法,在I 期临床试验中,给药组可显著降低患者的 ANGPTL3 和甘油三酯水平,并且维持疗效长达 16 周[53]。目前,Arrowhead 已经向美国 FDA 递交申请,启动 II 期临床试验,检验 ARO-ANG3 在血脂异常患者中的疗效。礼来公司研发的靶向 ANGPTL3 的 RNAi 疗法(LY3561774)目前正处于 I 期临床试验中。
3.3 基因编辑
利用基因编辑技术可从 DNA 层面上抑制 ANGPTL3进行降脂治疗。基因编辑利用了在基因组所需位置产生双链DNA 断裂的工具,2013年初 CRISPR-Cas9 系统作为基因组编辑工具的引入是基因组编辑领域的一个分水岭[51,54-58],而后对 CRISPR-Cas9 进一步改造,使其可以直接改变DNA 序列中的特定核苷酸,而不产生双链断裂,也不需要修复模板,即碱基编辑技术[59-63]。使用碱基编辑器已成功将angptl3突变导入小鼠肝细胞,使得小鼠体内甘油三酯和胆固醇水平均有明显下降[64]。现有研究团队进一步开发了脂质纳米颗粒输送平台,可携带 Cas9 信使 RNA 和引导RNA,用于基于 CRISPR-Cas9 的 ANGPTL3 体内基因组编辑[65]。Verve Therapeutics 公司研制的利用腺嘌呤碱基编辑(ABE)技术,可以直接修改肝脏细胞的基因组,将ANGPTL3 表达完全关闭,达到“一次治疗,终生获益”的效果。在临床前非人灵长类动物试验中,靶向 ANGPTL3 基因的体内碱基编辑疗法能够将动物血液中的 ANGPTL3 蛋白表达水平降低 95%,将血液中的甘油三酯水平降低64%。基因编辑技术是一项具有前景的治疗手段,但是若在人体中实现治疗还需解决伦理问题,并进行安全认证。
4 总结
在过去的 20 多年中,我们逐渐认识了 ANGPTL3 的重要性,从简单的调节 TG 到成为血脂异常及治疗动脉粥样硬化的一个重要靶点,尽管还有很多问题不清晰,例如ANGPTL3 调节 LDL-C 的机制,但相信随着研究的深入,以 ANGPTL3 作为靶点研发的抑制剂对于改善脂质代谢紊乱及降低心血管疾病的风险将会发挥重要的临床价值。
猜你喜欢
湖北农业科学(2022年11期)2022-07-18
锦州医科大学报(2021年8期)2021-11-18
家庭医药(2021年6期)2021-07-23
家庭科学·新健康(2020年6期)2020-07-06
百姓生活(2020年12期)2020-05-06
科学导报(2019年45期)2019-09-23
保健与生活(2019年23期)2019-09-10
祝您健康(2018年12期)2018-11-27
分析化学(2018年1期)2018-01-18
中文信息(2017年2期)2017-04-13
