
线纹海马肠道菌群结构与功能对迟钝爱德华氏菌(Edwardsiella tarda)感染的响应特征研究
2022-03-31 01:01张乐乐邹强田雅楠吕春晖郑诗怡姜广峻高龙坤侯玉平王凯
热带海洋学报 2022年2期
张乐乐, 邹强, 田雅楠, 吕春晖, 郑诗怡, 姜广峻, 高龙坤,侯玉平, 王凯
1. 鲁东大学农学院, 山东 烟台 264025;
2. 烟台市科技创新促进中心, 山东 烟台 264003;
3. 鲁东大学生命科学学院, 山东 烟台 264025
海马(Hippocampusspp.)具有很高的经济、观赏和药用价值(刘国信 等, 2006), 是我国二级保护动物、国际华盛顿公约指定保护物种(Lourie et al,1999)。作为海洋生态环境的旗舰物种(Lourie et al,2004; Cohen et al, 2017), 野生海马资源已濒临枯竭(Ternes et al, 2016)。加之中药材市场对海马的需求量持续增加, 导致海马价格持续攀升。我国广东、广西、福建、海南、浙江和山东等省份都出现了海马养殖的报道(吕军仪 等, 2001; 邓钢 等, 2005; Lin et al, 2008, 2010; Li et al, 2015; Wang et al, 2016,2017), 养殖热潮逐渐兴起。
过去30 年, 尽管全世界水产养殖业有了很大的提升, 但细菌性疾病的发生限制了集约化养殖的进一步发展(Pérez-Sánchez et al, 2018)。海马养殖业也面临同样的挑战。养殖过程中, 尤其在越冬后和交配间隔期, 细菌感染诱发的肠炎是导致亲本和幼龄海马大量死亡的主要原因(Wang et al, 2016; Qin et al,2017, 2018), 严重影响海马的交配效率及成活率,最终导致产量和经济效益下滑。因此, 细菌性肠炎病原的分离、鉴定, 发病机理的探索, 以及构建相应的防控体系, 成为提升海马工厂化养殖效率的关键环节。
目前已报道的海马细菌性病原超过20 种, 其中大部分能够诱发海马肠炎。上述病原多发现于水族养殖环境中, 发病机制尚不明确(Balcázar et al, 2010;Harikrishnan et al, 2010; LePage et al, 2012)。我国作为世界上最早尝试海马工厂化繁育且现有养殖量最大的国家, 近年来才刚开展细菌疾病研究, 尤其是细菌性肠炎的相关研究(Li et al, 2015; Qin et al,2018; Wang et al, 2016, 2020c; Shao et al, 2019)。目前只是对海马工厂化养殖过程中的细菌病原种类和危害有了初步了解, 发病机制以及防治方法方面的研究仍然十分缺乏。我们前期从海马肠道中分离出一种细菌性肠炎致病菌——迟钝爱德华氏菌(Edwardsiella tardaYT1), 并构建了海马细菌性肠炎的研究模型(Wang et al, 2020c), 为探究病原对海马肠道菌群的影响提供了研究基础。
本研究拟以国内养殖量最大的线纹海马(Hippocampus erectus)为研究对象, 利用上述研究模型, 并结合16S rDNA 高通量测序技术, 探讨E.tardaYT1 侵染对肠道菌群结构、多样性、丰度及功能的影响, 为海马细菌性肠炎致病机制的深入探索提供理论依据。
1 试验材料与方法
1.1 试验动物
所有线纹海马(H. erectus)取样按照实验动物护理和使用指南的要求进行操作。试验方案得到了鲁东大学伦理研究委员会的批准。五月龄健康线纹海马(体重3.86±0.023g、体长9.58±0.037cm) 购自威海银泽生物科技股份有限公司, 在鲁东大学农学院海洋生物养殖中心的智能海水养殖系统室中继续饲养,并以塑料植物作为附着基。养殖系统是具有机械和生物过滤、紫外线灭菌、蛋白分离和连续充气功能的中央循环系统。养殖用水盐度为31.5‰±0.5‰, 温度为25.5±0.5℃, pH 为8.2±0.1。为避免交配对试验结果的影响, 试验前将雌、雄海马分开暂养2 周。每天投喂冰冻糠虾3 次(08:00, 12:00 和16:00), 喂食后2h 吸出残留的饲料和粪便。
1.2 病原人工感染
注射试验共使用20 只海马, 将海马分成4 组,每组包含5 只海马。将活化后长至对数生长期的E.tardaYT1 离心, 用鱼类专用生理盐水 PSS(physiological saline solution)稀释重悬制成浓度为107个·mL-1的菌液备用。将100μL PSS (对照组: Con)和制备好的菌液(胁迫组: ET)分别注射到海马的腹腔后, 将其放回养殖缸中, 关闭循环水系统, 此后每天早上投饵2h 后将海马转入新养殖缸中饲养, 重复上述操作直至第21 天取样前。
1.3 样本采集
取样前一晚停止投饵, 取样前时先用鱼安定tricaine methanesulfonate (MS-222) (Sigma-Aldrich,Australia)麻醉处理海马, 随后用解剖刀和镊子取出完整肠道并将其置于冻存管中, 迅速放入液氮中。取样结束后, 将所有冻存管从液氮中取出, 立即放入提前准备好的干冰中, 打包寄往上海美吉生物医药科技有限公司进行后续测序。每组实验数据均进行5 次有效生物重复。
1.4 DNA 提取和测序
海马肠道样本在冰上做均质处理, 使用十六烷基三甲基溴化铵(hexadecyltrimethylammonium bromide, CTAB)法, 参考李焕宇等(2017)的具体步骤, 提取海马肠道菌群基因组DNA。用1%琼脂糖凝胶电泳分析总基因组DNA 的质量和完整性, 并借助超微量分光/荧光光度计(DS-11FX/FX+, 美国)鉴定DNA 质量。使用338F (5′-ACTCCTACGGGAG GCAGCAG-3′)和806R (5′-GGACTACHVGGGTWT CTAAT-3′)引物扩增所有样品的16S rDNA 的V3 和V4 区。PCR 产物使用QuantiFluor™-ST 蓝色荧光定量系统(Promega 公司, 北京)进行定量和纯化, 之后按照每个样本的测序量要求, 进行相应比例的混合。PCR 产物混合物用凝胶提取试剂盒纯化。根据操作说明, 使用 TruSeqTMDNA 样品制备试剂盒(Illumina, USA)生成测序文库。上机测序前, 需要先采用Agilent High Sensitivity DNA Kit 对文库在Agilent Bioanalyzer 上进行质检。然后采用Quant-iT PicoGreen dsDNA Assay Kit 在Promega QuantiFluor荧光定量系统上对文库定量。最后, 在 Illumina-HiSeq 平台上对该文库进行测序, 并产生250bp 的成对末端读序。
1.5 生物信息学和统计分析
采用Illumina-HiSeq 平台对群落DNA 片段进行双端(Paired-end)测序。所有读序都根据其唯一的条形码进行排序。使用FLASH 软件读取合并重叠的双端配对序列(Magoč et al, 2011)。使用QIIME 软件包对序列分解和质量筛选(Caporaso et al, 2010)。使用UCHIME 算法去除嵌合体序列(Edgar et al, 2011)。使用QIIME 软件, 调用UCLUST 这一序列比对工具(Edgar, 2010), 对前述获得的序列按97%的序列相似度进行归并和分类单元 OTU (operational taxonomic units)划分, 去除丰度值低于全体样本测序总量0.001%的OTU (Bokulich et al, 2013), 并将去除了稀有OTU 的序列作为该OTU 的代表序列进行进一步分析。
试验数据均以平均值±标准差形式呈现, 并采用SPSS 23.0 进行独立样本T检验分析,p<0.05 被认为是有显著性差异,p<0.01 被认为是有极显著性差异。使用QIIME 软件, 对OTU 丰度矩阵中每个样本的序列总数在不同深度下随机抽样, 以每个深度下抽取到的序列数及其对应的OTU 数绘制显示物种丰富度的稀疏曲线。使用QIIME 软件包对群落丰富度指数(ACE 和Chao)、多样性指数(Shannon 和Simpson)和覆盖率(Good’s coverage)的值进行计算(Caporaso et al, 2010)。基于Weihted-UniFrac 距离矩阵计算的β多样性指数, 采用主坐标分析法(PCoA)(Lozupone et al, 2006), 比较了健康海马和胁迫组海马的肠道菌群结构。使用Pearson 相关分析以评估肠道菌群与功能预测之间的关系。
2 结果
2.1 16S rDNA 测序概述及E. tarda YT1 侵染对海马肠道菌群α 和β 多样性的影响
从6 个测序样品中总共得到的有效序列数目为399424 条, 每个样品平均含有28918 条。其中有效碱基数有169130238bp, 每个样品的序列平均长度为423.44bp (表1)。这些序列以97%的相似性水平聚集到906 个OTUs , 分别属于25 门、55 纲、148目、260 科、483 属(图1 a)。6 个样品的OTUs 稀释曲线在右侧变平坦, 趋于饱和(图1b)。

图1 Edwardsiella tarda YT1 侵染前后海马肠道菌群16S rDNA 测序分析a. OTU 统计; b. 基于OTUs 绘制的稀释曲线。Con 为PBS 注射对照组; ET 为E. tarda YT1 胁迫组Fig. 1 Basic analysis of 16S rDNA sequencing of intestinal microbiota during Edwardsiella tarda YT1 infection of Hippocampus erectus. (a) OTU statistics; (b) Rarefaction curves of OTUs of intestinal microbiota of sampled seahorses
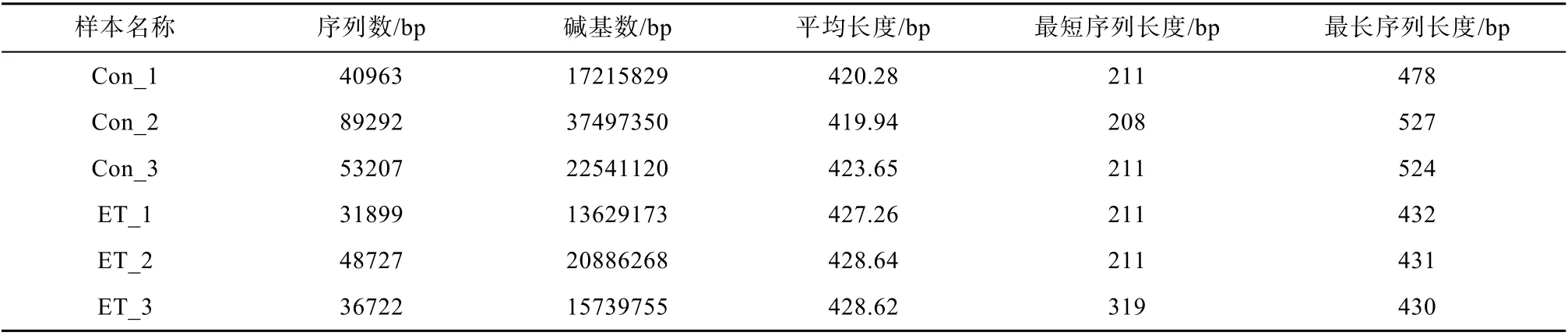
表1 样本信息统计表Tab. 1 Statistics of sequencing data from each sample
如图2 所示, 对照组共鉴定出635 个OTU, 胁迫组鉴定出522 个OTU, 两组共有的OTU 有251个。β多样性分析发现, 对照组与胁迫组海马肠道菌群组成结构区别明显。α多样性指数分析显示, 两组间Shannon 和Simpson 指数存在显著差异(p<0.05),ACE 和Chao 两个指数没有显著性差异(表2)。

表2 迟钝爱德华氏菌的侵染对海马肠道菌群α 多样性的影响Tab. 2 Effects of Edwardsiella tarda YT1 infection on α diversity of intestinal microbiota of lined seahorses
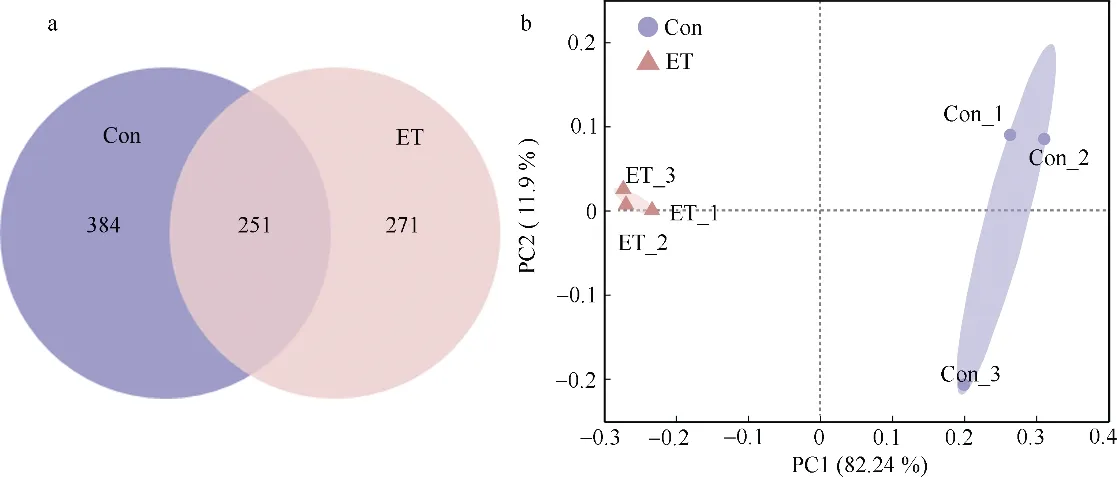
图2 Edwardsiella tarda YT1 侵染对海马肠道菌群组成结构的影响a. OUT 水平Venn 图; b. 肠道菌群的PCoA 加权分析。Con 为PBS 注射对照组; ET 为E. tarda YT1 胁迫组Fig. 2 Effect of Edwardsiella tarda YT1 infection on composition and structure of intestinal microbiota of lined seahorse. (a)The diagram of Venn at OUT level; (b) PCoA of weighted UniFrac distance matrices of intestinal microbiota
2.2 E. tarda YT1 侵染对门和属水平海马肠道菌群组成和相对丰度的影响
如图3 所示, 对照组和胁迫组肠道中共检测到12个相对丰度>0.1%的门。对照组中变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、厚壁菌门(Firmicutes)和拟杆菌门(Bacteroidetes)相对丰度均超过4%, 为优势菌门。胁迫组中变形菌门(Proteobacteria)相对丰度达到97%, 占绝对优势。与对照组相比, 胁迫组中变形菌门(Proteobacteria)显著增多(p<0.05), 而放线菌门(Actinobacteria)、厚壁菌门(Firmicutes)和拟杆菌门(Bacteroidetes)显著减少(p<0.05)。
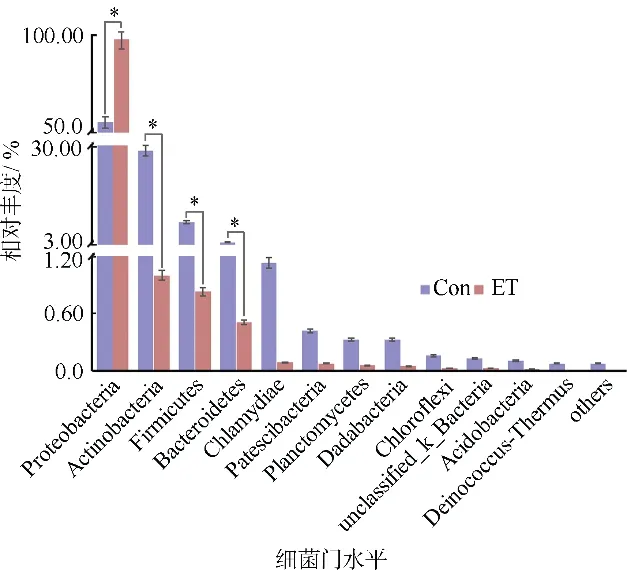
图3 Edwardsiella tarda YT1 侵染对海马肠道菌群(门水平)相对丰度的影响Con 为PBS 注射对照组; ET 为E. tarda YT1 胁迫组。*表示两组有显著性差异(p<0.05)Fig. 3 Effect of Edwardsiella tarda YT1 infection on relative abundance of intestinal microbiota (relative abundance > 0.1%) at phylum level of lined seahorse (* p <0.05)
通过属水平肠道菌群的相对丰度对比发现: 对照组和胁迫组中相对丰度超过1%的属共有22 个(图4); 对照组中相对丰度超过1%的属有20 个, 相对丰度由高到低依次为嗜冷杆菌属(Psychrobacter)、分枝杆菌属(Mycobacterium)、罗氏菌属(Rothia)、动球菌属(Planococcus)、青枯菌属(Ralstonia)、谷氨酸杆菌属(Glutamicibacter)、球菌属(Macrococcus)、鲁杰氏菌属(Ruegeria)、微小杆菌属(Exjguobacterium)、阿赫兰氏菌属(Ahrensia) 、 别样玫瑰变色菌属(Aliiroseovarius) 、 伯克氏菌属(Burkholderia-Caballeronia-Paraburkholderia) 、 不动杆菌属(Acinetobacter)、金黄杆菌属(Chryseobacterium)、鼠尾菌属(Muricauda)、链球菌属(Streptococcus)、未分类-衣原体(unclassified-Chlamydiales)、未标注-生丝微菌科(norank-Hyphomicrobiaceae)、葡萄球菌属(Staphylococcus) 和未标注- 根瘤菌(norank-Rhizobiaceae); 胁迫组中相对丰度超过1%的属有9个, 相对丰度由高到低依次为爱德华氏菌属(Edwardsiella) 、 弧菌属(Vibrio) 、 嗜冷杆菌属(Psychrobacter)、分枝杆菌属(Mycobacterium)、谷氨酸杆菌属(Glutamicibacter)、罗氏菌属(Rothia)、鲁杰氏菌属(Ruegeria)、鼠尾菌属(Muricauda)和未标注-根瘤菌(norank-Rhizobiaceae)。

图4 Edwardsiella tarda YT1 侵染对海马肠道菌群(属水平)相对丰度的影响内、外圈分别显示肠道菌群在属水平的相对丰度与名称。Con 为PBS 注射对照组; ET 为E. tarda YT1 胁迫组Fig. 4 Comparison of intestinal microbiota between healthy and diseased seahorses at genus level (relative abundance > 1%)by circular representation. The inner and outer circular diagrams show the relative abundance of intestinal microbiota at genus level and name, respectively
通过属水平肠道菌群相对丰度的差异分析, 发现7 个相对丰度较高且病原侵染前后相对丰度差异明显的菌属有爱德华氏菌属、嗜冷杆菌属、动球菌属、谷氨酸杆菌属、罗氏菌属、青枯菌属与动球菌属(图5a)。定量分析显示, 胁迫组中爱德华氏菌属的相对丰度极显著增多(p<0.01), 而冷杆菌属与罗氏菌属呈极显著性降低(p<0.01), 动球菌属与球菌属呈显著性降低(p<0.05)(图5b)。

图5 Edwardsiella tarda YT1 侵染对肠道中优势菌群相对丰度的影响a. 基于线性判别分析效应(LEfSe)绘制的海马肠道微生物群落的物种群落结构图, 图中字母指代菌属见图b。不同颜色节点表示在对应组别中显著富集, 且对组间差异存在显著影响的微生物类群; 淡黄色节点表示在不同分组中均无显著差异或对组间差异无显著影响的微生物类群。节点大小显示了物种的丰度; b. 差异明显的属的定量分析。*表示p<0.05, **表示p<0.01。Con 为PBS 注射对照组;ET 为E. tarda YT1 胁迫组Fig. 5 Comparison of dominant intestinal microbiota between healthy and diseased seahorses. (a) Species community structure diagram of gut microbiomes of the seahorse by linear discriminant analysis effect size (LEfSe); (b) Quantitative analysis of genus (* p<0.05, ** p<0.01). The node’s size displays the abundance of the species
2.3 E. tarda YT1 侵染对海马肠道菌群功能的影响
通过对比肠道菌群功能的差异发现, 对照组与胁迫组间差异最显著(p<0.05)的功能主要聚焦在细菌趋化性、鞭毛组装、ABC 转运蛋白、磷酸转运酶系统、核糖体、RNA 降解与核苷酸剪切修复、脂肪酸生物合成和脂多糖生物合成等方面(图6)。病原胁迫后, 细菌趋化性、鞭毛组装、ABC 转运蛋白、磷酸转运酶系统和脂多糖生物合成途径的活性显著上调(p<0.05), 而核糖体、RNA 降解、核苷酸剪切修复和脂肪酸生物合成途径的活性显著降低(p<0.05)。

图6 Edwardsiella tarda YT1 侵染对海马肠道菌群功能的影响Con 为PBS 注射对照组; ET 为E. tarda YT1 胁迫组Fig. 6 Functional differences of intestinal microbiota during Edwardsiella tarda YT1-induced enteritis in lined seahorses
通过对比海马肠道菌群的相对丰度与功能相关性分析发现, 病原菌和正常肠道优势菌群相对丰度的改变与肠道菌群功能活性的变化存在显著相关性(图7)。爱德华氏菌属的相对丰度与细菌趋化、鞭毛组装、ABC 转运蛋白、磷酸转运酶系统和脂多糖生物合成途径的活性显著正相关(p<0.05), 而与脂肪酸生物合成、核苷酸剪切修复、RNA 降解和核糖体的活性显著负相关(p<0.05); 动球菌属和谷氨酸杆菌属的相对丰度与核糖体、核苷酸剪切修复、RNA 降解和脂肪酸生物合成途径的活性显著正相关(p<0.05),与细菌趋化、鞭毛组装途径的活性极显著负相关(p<0.001); 嗜冷杆菌属的相对丰度与脂肪酸生物合成、核苷酸剪切修复途径的活性显著正相关(p<0.05),与脂多糖生物合成、ABC 转运蛋白和磷酸转运酶系统的活性极显著负相关(p<0.001); 罗氏菌属与ABC 转运蛋白的活性显著负相关(p<0.05); 青枯菌属的相对丰度与脂多糖生物合成、磷酸转运酶系统、细菌趋化和鞭毛组装途径的活性显著负相关(p<0.05); 球菌属的相对丰度与脂多糖生物合成、磷酸转运酶系统途径的活性存在显著负相关(p<0.05)。

图7 病原侵染过程中海马肠道优势菌属与功能的相关性分析红色代表优势菌属与功能呈正相关, 蓝色代表负相关。*表示p<0.05, **表示p<0.01, ***表示p<0.001。图上方的聚类分析表示肠道优势菌属共分为两大支, 其中Edwardsiella 为一支, 其他优势属为一支Fig. 7 Correlation between relative abundance of dominant genera and functions of intestinal microbiota during Edwardsiella tarda YT1-induced enteritis in lined seahorses (* p<0.05, ** p<0.01, *** p<0.001)
3 讨论与结论
肠道菌群结构对于宿主抵抗外源生物的侵染至关重要(Lozupone et al, 2012; Nowarski et al, 2017; Zhou et al, 2020)。鱼的胃肠道中定居着超过10 亿种以细菌为主的微生物, 与鱼类建立相互作用、相互影响的关系(Robertson et al, 2018)。当宿主与肠道菌群之间的动态平衡遭到破坏, 则会导致或促进疾病的发生(Hayakawa et al, 2016; Tran et al, 2018; Xiong et al,2019)。本研究中,E. tardaYT1 侵染能降低线纹海马肠道菌群OTU 水平的多样性, 并改变其组成结构(图2和表 2), 这与病原感染诱发大海马(Hippocampus kuda)、斑马鱼(Danio rerio)、草鱼(Ctenopharyngodon idellus)和圆口铜鱼(Coreius guichenoti)中细菌性肠炎的研究结果一致(Li et al, 2016; Yang et al, 2017; Tran et al, 2018; Wang et al, 2020b), 表明细菌病原侵染可以通过降低鱼类肠道菌群的多样性和改变其组成结构,从而打破肠道菌群的平衡。
变形菌门是鱼类中最丰富的细菌门, 其相对丰度的增加与肠道炎症和宿主免疫监管有关(Roeselers et al, 2011; Shin et al, 2015; Yang et al,2017; Tyagi et al, 2019)。厚壁菌门可以调节鱼类的免疫反应(Zhang et al, 2015)。拟杆菌门与碳水化合物代谢有关(Xu et al, 2003; Kamada et al, 2012)。而放线菌门是肠道菌群中最普遍的成员(Wu et al,2012a)。本研究中,E. tardaYT1 的侵染显著增加变形菌门相对丰度(p<0.01)的同时, 能显著减少放线菌门、厚壁菌门和拟杆菌门海马肠道固有菌群的相对丰度(p<0.05)(图3)。这与草鱼、铜鱼和斑马鱼患细菌性肠炎后门水平菌群丰度变化的结果一致(Li et al, 2016; Yang et al, 2017; Tran et al, 2018)。上述细菌病原同属变形菌门, 这可能是导致变形菌门相对丰度增加的主要原因。另有研究发现, 无乳链球菌(Streptococcus agalactiae)感染斑马鱼显著增加肠道梭杆菌门相对丰度的同时, 明显减少了变形菌门、厚壁菌门和放线菌门的相对丰度(Zhang et al,2019)。上述结果提示, 病原侵染诱发鱼类细菌性肠炎过程中, 致病菌相对丰度的增加可以抑制肠道内固有菌群的丰度, 打破菌群的动态平衡。
肠道核心菌群的丰度和功能对于维持鱼类内稳态和健康具有重要意义(Frank et al, 2007; Clemente et al, 2012)。目前已报道的鱼类肠道固有菌属有嗜冷杆菌属、芽孢杆菌属(Bacillus)、动球菌属、谷氨酸杆菌属、不劳蒂亚属(B l a u t i a)和乳球菌属(Lactococcus)(Wu et al, 2012b; Liu et al, 2020; Wang et al, 2020b)。嗜冷杆菌属作为一种潜在的水产养殖益生菌, 可以提高鱼类的生长性能与消化酶活性,上调免疫相关基因的表达, 对抗细菌原体侵染(Caipang et al, 2010; Yang et al, 2011; Makled et al,2017); 动球菌属和谷氨酸杆菌属与代谢途径密切相关(Buschke et al, 2011), 其中动球菌属参与脂类合成途径, 该途径衍生的牛血清蛋白具有抗菌、免疫调节作用(Waghmode et al, 2020), 产生的生物表面活性剂可以抑制致病生物(Manikandan et al, 2017);谷氨酸杆菌属可以促进纤维素、半纤维素的消化,有助于提高宿主的消化和吸收功能(Wang et al,2020a)。本研究发现,E. tardaYT1 侵染显著增加爱德华氏菌属相对丰度的同时(p<0.01), 明显降低了海马肠道固有菌群中嗜冷杆菌属、动球菌属、谷氨酸杆菌属、罗氏菌属、青枯菌属与球菌属的相对丰度(图5), 提示上述菌属的丰度和功能可能对于维持海马肠道的内稳态具有重要作用。另有研究表明病原侵染大海马、斑马鱼以及珍珠龙胆石斑鱼肠道后,明显减少了嗜冷杆菌属、动球菌属、不劳蒂亚属、谷氨酸杆菌属、不动杆菌属(Acinetobacter)和发光杆菌属(Photobacterium)等肠道固有菌群的相对丰度(Li et al, 2016; Yang et al, 2017; Deng et al, 2020)。由上可知, 致病菌可能通过减少鱼类肠道固有菌群的丰度, 改变其结构与功能, 诱导菌群和功能失调,促进疾病发生(Xiong et al, 2015; Li et al, 2019; Liu et al, 2020)。
研究表明, 脂多糖生物合成与病原菌毒力相关因子有关(Yu et al, 2016); 细菌趋化性在动物-细菌间的相互作用和病原感染的各个阶段都发挥着至关重要的作用(Hammer et al, 2003; Williams et al,2007; Erhardt, 2016; Matilla et al, 2018)。ABC 转运蛋白与营养、维生素和代谢物的吸收有关(Petryszyn et al, 2018); 磷酸转运酶系统在调控运输、代谢和基因表达方面具有重要作用(Miyata et al, 2013;Rivera-Pérez et al, 2019), 在病原菌侵染过程中参与刺激生物膜的形成、聚集运动和诱导细菌的定植(Gao et al, 2019)。本研究中,E. tardaYT1 侵染后细菌趋化性、鞭毛组装、ABC 转运蛋白、磷酸转运酶系统和脂多糖生物合成功能活性显著上调, 而且上述功能活性与条件致病菌爱德华氏菌属的丰度呈显著正相关(p<0.05)(图4~图6), 这表明E. tardaYT1可能通过趋化作用、营养物质的竞争和毒力效应与肠道固有菌群竞争, 致使肠道菌群结构和功能的内稳态失衡, 促进疾病发生。肠道固有菌群能通过执行不同的功能抵御病原的侵染(Purchiaroni et al,2013)。核糖体参与免疫信号过程, 具有抗炎作用(Zhou et al, 2015); RNA 降解与疾病的监测密切相关(Sohrabi-Jahromi et al, 2019); 脂肪酸生物合成为各种代谢提供能量, 在病原体防御中起重要作用(Lim et al, 2017)。本研究中,E. tardaYT1 侵染后海马肠道固有菌群中发挥核心作用的嗜冷杆菌属、动球菌属和谷氨酸杆菌属的丰度与核糖体、RNA 降解、核苷酸剪切修复和脂肪酸生物合成功能活性显著正相关(p<0.05), 且均在病原侵染后明显下调, 提示上述核心菌群在海马肠道病原的监测和防御中发挥着重要的功能。本研究结果提示,E. tardaYT1 侵染可能通过自身的趋化作用、毒力效应和营养竞争等手段降低海马肠道菌群多样性, 改变肠道菌群的组成结构, 抑制海马肠道核心菌群的相对丰度和功能, 并诱发肠炎。综合分析、考量鱼类中已有的报道、海马自身特征, 以及安全和可持续发展等因素, 益生菌(probiotics)、益生元(prebiotics)、天然抗菌化合物(natural antimicrobial compounds)、细菌的群体感应(quorum sensing)和遗传育种(genetic breeding)等制剂和技术在海马工厂化养殖过程中细菌性疾病,尤其是细菌性肠炎防控中的应用潜力巨大。
综上所述, 本研究探讨了E. tardaYT1 感染对线纹海马肠道菌群的影响。结果发现, 与其他鱼类相似,E. tardaYT1 感染海马后可以从不同的水平改变肠道菌群的多样性、结构组成和丰度。究其原因,E. tardaYT1 可能一方面通过其丰度的增加, 另一方面通过其毒力、趋化和营养竞争作用的发挥, 诱导海马肠道菌群结构和功能的失调, 进而诱发肠炎。
猜你喜欢
安徽农业科学(2022年20期)2022-11-11
科学技术与工程(2022年26期)2022-11-01
河南医学研究(2022年19期)2022-10-19
中国农学通报(2022年14期)2022-06-01
分子诊断与治疗杂志(2022年4期)2022-05-30
油气田环境保护(2022年2期)2022-05-09
中国典型病例大全(2022年7期)2022-04-22
国际消化病杂志(2021年1期)2021-03-05
特别健康·下半月(2019年7期)2019-07-29
特别健康·下半月(2017年5期)2017-06-15
