
Identification of quantitative trait loci associated with resistance to Xanthomonas oryzae pv. oryzae pathotypes prevalent in South China
2022-03-30 08:51JilingLuQunlinLiChunchoWngMingmingWngDnZengFnZhngWenxueZhiYongliZhou
The Crop Journal 2022年2期
Jiling Lu, Qunlin Li, Chuncho Wng, Mingming Wng, Dn Zeng, Fn Zhng, Wenxue Zhi,*,Yongli Zhou,*
a National Key Facility for Crop Gene Resources and Genetic Improvement/Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, Beijing 100081, China
b Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing 100101, China
Keywords:Rice Bacterial blight Genome-wide association study CRISPR-Cpf1
ABSTRACT Bacterial blight(BB),which is caused by Xanthomonas oryzae pv.oryzae(Xoo),is an important rice disease responsible for significant yield losses.In the rice-growing regions of South China where BB outbreaks are common,the resistance of cultivars with BB resistance genes Xa4 and Xa21 has been lost because of rapid changes in the Xoo population structure and virulence. In this study, 421 diverse rice accessions were evaluated regarding their resistance to two Xoo strains,namely GD1358(C5)and IV,which are prevalent pathotypes in South China and overcame the resistance of Xa4 and Xa21, respectively. Using the 4.8mio filtered SNP dataset,we conducted a genome-wide association study,which identified 13 loci associated with BB resistance, including eight new quantitative trait loci (QTL) and five QTL harboring known BB resistance genes: Xa3/Xa26, xa5, Xa35(t), Xa36(t), Xa40, Xa43(t), and xa44(t). Intriguingly, a steep peak was detected on chromosomes 5 and 11.Six QTL including three new ones,were distributed on chromosome 11,whereas a new QTL qBB5.1 and a known QTL were detected on chromosome 5.Haplotype analyses indicated that the LOC_Os05g01610(OsPRAF2)gene within the qBB5.1 region,which encodes a PRAF protein, is associated with BB resistance. Furthermore, OsPRAF2 knockout lines generated using the CRISPR-Cpf1 system were significantly more resistant to Xoo strains than the wild-type plants. Our results provide researchers and breeders with useful information regarding QTL and gene resources,which may be relevant for developing new BB-resistant rice cultivars.
1. Introduction
As a staple cereal crop,rice(Oryza sativa L.)is an important food source for approximately half of the global population[1,2].Bacterial blight (BB), which is caused by Xanthomonas oryzae pv. oryzae(Xoo),is a major threat to rice production worldwide, especially in Asia,where 90%of the global rice yield is produced and consumed[3]. This disease can decrease rice yield by 20%–30%, but in severe cases, the yield loss can reach 50% [4]. In the rice-growing regions of South China, outbreaks of BB are common because of the subtropical monsoon climate,making it a serious disease constraining rice production[5,6].The rice–Xoo interaction follows the classical gene-for-gene model [7]. Breeding and deploying resistant cultivars is the most economical and eco-friendly strategy for controlling BB [8,9].
To date, 46 disease resistance-related genes Xa1–Xa46(t) have been identified in different chromosomal regions[10–14],of which Xa1, Xa3/Xa26, Xa4, xa5, Xa7, Xa10, xa13, Xa21, Xa23, xa25, Xa27,and xa41(t) have been cloned [15–27]. However, because these genes tend to confer relatively weak and narrow-spectrum disease resistance, only a few dominant genes that provide broadspectrum disease resistance, including Xa4, Xa21, Xa23, and Xa39,have been widely used in rice breeding programs [18,22,26,28].The narrow genetic basis of BB resistance in rice cultivars has resulted in a dramatic increase in the frequency of Xoo pathotype V in South China [5]. Furthermore, Xoo strains virulent to rice plants carrying Xa21 have been isolated in the Philippines, Korea,India, and China [29–31]. A pathotype virulent to plants carrying Xa23 was recently detected in Guangxi,South China[6].Therefore,new genes and quantitative trait loci(QTL)conferring resistance to Xoo pathotypes prevalent in South China will need to be identified.
During the last decade, genome-wide association studies(GWAS)have been widely conducted to decipher the genetic basis of complex traits in diverse varieties and to identify the causative loci underlying these traits [32]. For example, 56 and 27 QTL for resistance to Magnaporthe oryzae isolates from Hunan province and Yunnan province,respectively,in China were identified in rice[33,34]. Additionally, in an earlier study involving 285 rice accessions and nine Xoo strains from the Philippines,new resistance loci were detected on chromosomes 6, 9, 11, and 12 [35]. Two major loci for BB resistance, qBLB11.1 and qBLB5.1, were identified using a rice multi-parent advanced generation inter-cross (MAGIC) plus population[36].Moreover,Xa43(t)and seven other QTL associated with resistance to Xoo were identified in different MAGIC populations [11,37]. Using rice accessions from the 3000 Rice Genomes Project(3K RGP)[38,39],QTL and candidate genes associated with several traits, including resistance to BB and sheath blight, were detected [40–43].
In the present study,421 accessions from the 3 K RGP were evaluated regarding their resistanceto two prevalent Chinese Xoo pathotypes collected from South China. The accession genotypes were obtained from the 3K RG 4.8mio filtered SNP dataset. On the basis of the rice BB resistance phenotypes and high-density SNP genotypes,13 genetic loci associated with BB resistance were identified.Notably, we revealed that the PRAF (PH, RCC1 and FYVE) gene LOC_Os05g01610 (OsPRAF2) within qBB5.1 negatively regulates BB resistance.This study provides information regarding valuable rice germplasm potentially useful for breeding BB-resistant varieties and for clarifying the molecular basis of BB resistance.
2. Materials and methods
2.1. Plant and bacterial materials
The diverse panel comprising 421 rice accessions from the 3K RGP used in this study included 246 xian (XI), 75 geng (GJ), 15 admixture (Admix), 81 cA (Aus), and 4 cB (Bas) accessions [39].The seeds of all samples were first sown in a seedling nursery,after which 30-day-old seedlings were transplanted to a quarantine area at Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, Beijing, China. A field trial was completed using a randomized complete block design with two replicates.For each replicate, the accessions were grown in 1.2-m rows, with 20 cm separating rows and eight plants per row. Standard local agronomic practices, but with no bactericide applications, were used throughout the growing season.
Two representative strains of Xoo pathotypes prevalent in South China, IV (pathotype IV) and GD1358(C5, pathotype V) [31], were used for investigating BB resistance. Specifically, C5 and IV can overcome the resistance conferred by Xa4 [44] and Xa21 [31],respectively.
2.2. Phenotypic evaluation of BB resistance
The BB resistance of rice plants was evaluated at the maximum tillering stage. As previously described, plants were inoculated using the scissor-cutting leaf method and examined 3 weeks later when the lesion length (LL) was stable [28,45]. The average LL of two replicates was used to construct the phenotype matrix for subsequent analyses.On the basis of the LL,rice accessions were classified as follows[42]:resistant(R):LL <5 cm;moderately resistant(MR): 5 cm ≤ LL < 10 cm; moderately susceptible (MS):10 cm ≤LL <15 cm; or susceptible (S): LL ≥15 cm.
2.3. Principal component analysis, kinship, and genetic structure
A total of 249,632 independent SNPs were obtained from the 3 K RG 4.8mio filtered SNP dataset [46] using the following filtering parameters: missing rate > 0.1, minor allele frequency(MAF)<0.05,and‘‘indep-pairwise 50 10 0.5”[47].A principal component analysis (PCA) was performed using smart PCA in the EIGENSOFT program [48]. Genetic relationships between rice accessions were estimated using the Balding–Nichols method and a kinship matrix was generated[49].Rice accession population structures were predicted using the ADMIXTURE program, and K values were set from 2 to 9 to determine the lowest coefficient of variation [50].
2.4. Genome-wide association mapping
A total of 3,080,542 SNPs in the 421 accessions were obtained from the 3 K RG 4.8mio filtered SNP dataset by filtering SNPs(missing rate >0.1 and MAF <0.05)using PLINK[47].Applying principal components and kinship as covariates,we conducted a GWAS with SNP genotypes and phenotypes using the Efficient Mixed-Model Association eXpedited (EMMAX) program[51].The whole population and subgroups with more than 200 accessions were analyzed separately for the GWAS. After filtering SNPs on the basis of the missing rate and MAF,3,080,542 and 2,088,873 SNPs were retained for the GWAS of the whole population and XI population, respectively. The P-value threshold for determining significance was estimated using the GEC software [52]. Manhattan and quantile–quantile plots were drawn using the R package CMplot (https://github.com/YinLiLin/R-CMplot).
A region with more than two significant SNPs in the linkage disequilibrium(LD)block was considered to be a QTL associated with BB resistance,and the SNP with the minimum P-value within each QTL was designated as the lead SNP [43,53].The LD block referred to the continuous region closely linked to the lead SNP (r2≥0.6),and the LD heat map was constructed using the LDheatmap package in the R environment[54,55].The SNPs within QTL were annotated on the basis of the ‘‘Effect of 29mio biallelic SNPs on Rice Genome Annotation Project rel 7 gene models” (https://snp-seek.irri.org).The haplotype analysis of candidate genes was conducted and the phenotypes of the major haplotypes (at least 10 accessions) were analyzed using Duncan’s multiple comparisons test[56].
Regarding the xa5 allele,we amplified xa5 fragments containing the two reported functional SNPs using primers xa5_1F and xa5_1R(Table S1). Approximately 50 ng genomic DNA was included in a 15-μL polymerase chain reaction (PCR), which was completed using 7.5 μL 2× Rapid Taq Master Mix (Vazyme Biotech Co., Ltd.,Nanjing,China)and 0.6 μL each primer(10 μmol L-1).The PCR conditions were as follows:95°C for 3 min;35 cycles of 95°C for 15 s,53 °C for 15 s, and 72 °C for 20 s; 72 °C for 5 min. To digest the amplified fragments, the PCR products (10 μL) were mixed with 2 μL 10× NEBuffer, 0.5 μL BsrI (5 U), and 7.5 μL ddH2O and then incubated at 65 °C for 4 h. The digested products were analyzed on 2.5% agarose gels in 1× TAE buffer. The accessions with one 299 bp fragment were considered to contain the xa5-resistance allele, whereas accessions with two fragments (199 and 100 bp)were considered to contain the xa5-susceptibility allele [57].
2.5. Construction of mutants mediated by CRISPR-Cpf1
The CRISPR-mediated gene editing system CRISPR-Cpf1 was used to generate candidate gene LOC_Os05g01610 (OsPRAF2)knockout mutants [58]. The target sequence and primers were designed and the transgenic mutant lines were analyzed using CRISPR-GE(http://skl.scau.edu.cn/).The primers used in this study are listed in Table S1.The CRISPR vector LbCpf1 carrying the target gRNA was transformed into Nipponbare rice plants via a previously described Agrobacterium-mediated transformation method [59].
3. Results
3.1. Genotypes and population structures of rice accessions
A total of 3,080,542 high-quality SNPs were obtained, with a missing rate less than 10% and a MAF greater than 5%. An ultradense SNP genetic map was constructed (Fig. S1), in which SNPs were evenly distributed on each chromosome, with chromosomes 9 and 1 being the shortest and longest, respectively. More specifically, the number of SNPs on an individual chromosome ranged from 182,918 (chromosome 9) to 391,324 (chromosome 1). The average distance between SNPs in the whole genome was 121 bp, ranging from 110 bp (chromosome 8) to 138 bp (chromosome 4) (Table S2).
The diverse rice panel in this study comprised 421 accessions from 58 countries, with varieties from India, Bangladesh, and the Philippines representing approximately 50% of the accessions(Fig. S2A). Moreover, of the included accessions, 58.43%, 17.81%,3.56%, 19.24%, and 0.95% were from the XI, GJ, Admix, cA, and cB subgroups, respectively (Fig. S2B). The PCA revealed three major subgroups, with the genetic variation explained by the first two principal components (Fig. 1A). Additionally, the heat map of the kinship was consistent with the three major subgroups, except for Admix and cB, which had relatively few accessions (Fig. 1B).The lowest coefficient of variation was generated when K was set to 3,indicating that the rice accessions could be divided into three major subgroups(Fig.1C).These results suggested that the diverse rice accessions in the panel had clear population structures and were suitable for the GWAS.
3.2. Evaluation of the resistance of the rice accessions to two Xoo strains
Two representative highly virulent prevalent Chinese Xoo strains (C5 and IV) from South China were selected to investigate the BB resistance of the rice accessions. The LL distribution varied considerably for the two Xoo strains, and the average LL was greater for the accessions inoculated with C5 than for the accessions inoculated with IV,which suggests that the panel was appropriate for screening resistant cultivars and for performing a GWAS to identify genetic loci (Fig. 2A). Overall, the number of resistant accessions (LL <5 cm) was approximately 2.5-times higher for IV than for C5. Additionally, more accessions were susceptible(LL ≥15 cm)to C5 than to IV,which implies that C5 was more virulent than IV (Fig. 2B). Of the inoculated accessions, 61 and 158 were highly resistant to C5 and IV, respectively. Moreover, 48 accessions were highly resistant to both strains,including 35 from Bangladesh,8 from India,3 from the Philippines,1 from Nepal,and 1 with an unknown origin; the cA subgroup accounted for about 85% of these accessions (Table S3).
A comparison of the BB resistance of rice accessions in different subgroups revealed that 2.44%, 16.00%, 13.33%, 50.62%, and 0% of the XI, GJ, Admix, cA, and cB accessions were resistant to C5,respectively (Fig. 2C), whereas 43.90%, 2.67%, 13.33%, 56.79%, and 0% of the XI, GJ, Admix, cA, and cB accessions were resistant to IV, respectively (Fig. 2D). These results indicated that cA was the most resistant subgroup, which suggests that it may be useful for breeding BB-resistant rice.

Fig.1. Population structure of 421 rice accessions.(A)Principal component analysis of accessions based on the first two components.(B)Heat map of the kinship matrix.(C)Distribution of the estimated subpopulation components for each accession determined by ADMIXTURE.

Fig.2. Evaluation of resistance to C5 and IV.(A)Violin plot of the lesion length among 421 accessions.(B)Number of accessions with different resistance reactions to the two strains. Resistance of five subgroups to C5 (C) and IV (D).
3.3. Identification of genetic loci associated with BB resistance
On the basis of the independent markers after a Bonferroni correction at the 0.05 significance level, P-values of 1.05E-7 and 3.20E-6 were set as the significance thresholds for the whole population and the XI population,respectively. A total of 360 and 419 SNPs in the whole population were associated with resistance to C5 (Fig. 3A) and IV (Fig. 3B), respectively. Interestingly, when the GWAS was performed for the XI population, only two significant SNPs were detected for C5(Fig.S3A),whereas 726 significant SNPs were detected for IV (Fig. S3B). Significant SNPs were distributed on all chromosomes, except for chromosomes 7 and 12, and were present in 13 QTL according to the QTL classification. The QTL related to BB resistance were designated as qBBN.X,where N refers to the chromosome and X represents the order on the same chromosome. Eight and eight loci were separately detected in the whole and XI populations, respectively, with three loci (qBB11.4,qBB11.5, and qBB11.6) simultaneously identified in both populations (Table 1). Notably, of the seven and ten loci associated with resistance to C5 and IV, respectively, four loci (qBB5.1, qBB5.2,qBB11.5,and qBB11.6)mediated resistance to both strains(Table 1),which implies that these loci might confer multifunctional resistance. Among the 13 QTL, five were co-located or closely linked to previously mapped genes or QTL in different populations,whereas the other eight loci were new (Table 1). Additionally,two strong association signals were detected on chromosomes 5 and 11(Fig.3A,B).Two QTL(one new)were identified on chromosome 5, whereas six QTL (three new) were detected on chromosome 11 (Table 1).
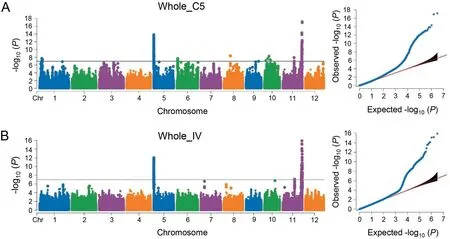
Fig. 3. Manhattan and quantile–quantile plots for the GWAS of the resistance to C5 (A) and IV (B) in the whole population.
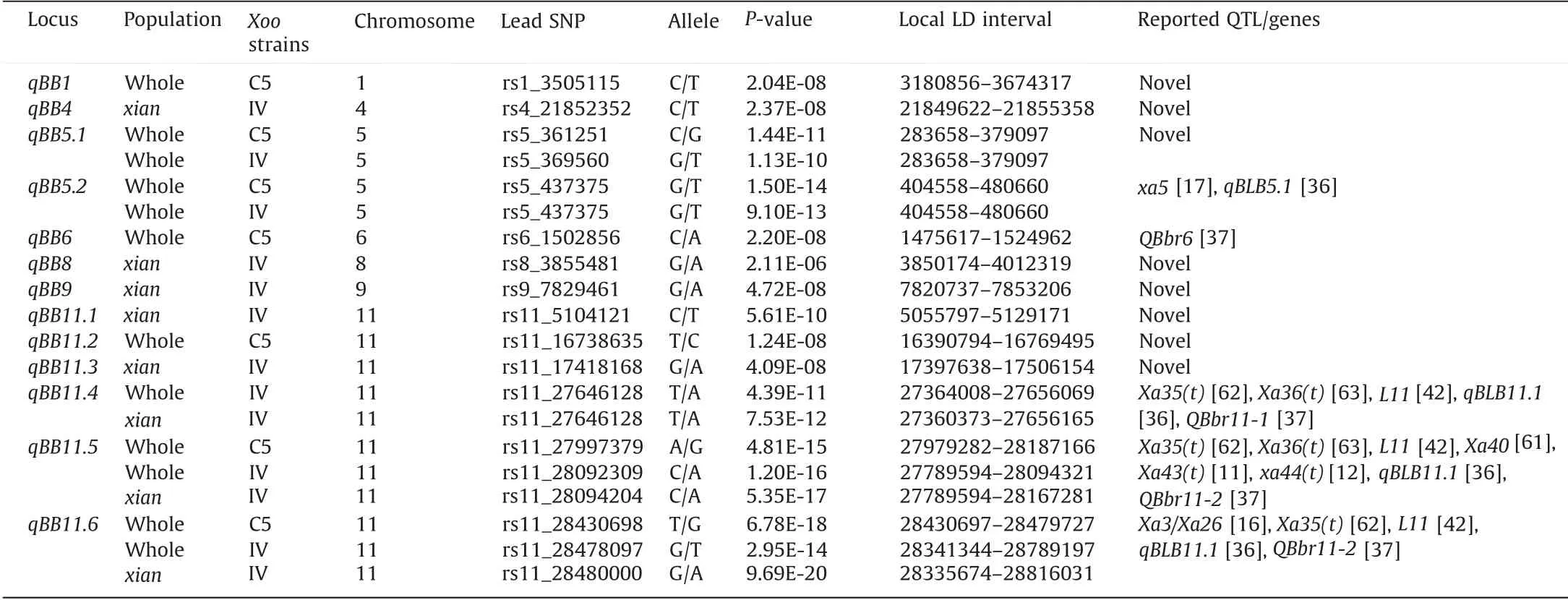
Table 1 Genetic loci associated with bacterial blight resistance identified in the genome-wide association study.
Regarding chromosome 5, qBB5.1 and qBB5.2 conferred resistance to both C5 and IV in the whole population. Intriguingly, the broadly effective resistance gene xa5 was located in the qBB5.2 region[17].On the basis of the reported functional polymorphisms in the second exon of LOC_Os05g01710 (xa5), we performed an association analysis to validate the regulatory effects of this gene on the resistance to C5 and IV. For LOC_Os05g01710, the 421 rice accessions were classified into two distinct haplotypes, with 363 accessions carrying Hap1 and 53 accessions carrying Hap2(Fig. S4A). Accessions with Hap2 had shorter lesions than accessions with Hap1 in response to infection by C5 and IV (Fig. S4B),which suggests that Hap2(i.e.,xa5)was the favorable allele conferring resistance to highly virulent Xoo strains. Furthermore, we tested for the presence of the xa5-resistance allele in the 48 rice accessions highly resistant to C5 and IV using the reported functional marker (xa5_1F/xa5_1R) and BsrI digestion [57]. The results indicated that with the exception of IRIS_313-8265, IRIS_313-10366, IRIS_313-10920, and IRIS_313-11979, the accessions contained the xa5-resistance allele with the two functional SNPs (A and G)at 437,499 and 437,500 bp of chromosome 5.However,rice plants with only xa5 were reportedly susceptible to Xoo strains carrying the TAL effector gene pthXo1[60].Accordingly,the pyramiding of xa5 with other genes or QTL is necessary for protecting plants against multiple Xoo strains. Therefore, the new QTL qBB5.1, which is associated with resistance to both C5 and IV,may include new resistance genes and should be explored more precisely.
3.4. Candidate gene prediction
The new locus qBB1, which was associated with the resistance to C5, was mapped to a 3.18–3.67 Mb interval on chromosome 1.This interval included significant SNPs rs1_3285444 and rs1_3505115 in LOC_Os01g06920 and LOC_Os01g07400, respectively. According to the annotations determined by the Rice Genome Annotation Project, LOC_Os01g06920 encodes the SlVe1 disease resistance protein precursor, which includes a leucinerich repeat domain. The protein encoded by LOC_Os01g07400 contains a WD40 repeat-like domain,and was predicted to participate in responses to extracellular stimuli.
The steep peak detected for chromosome 5 was divided into two QTL(qBB5.1 and qBB5.2)associated with the resistance to both C5 and IV. The lack of previously reported BB resistance-related genes in the candidate region of qBB5.1 (283,658–379,097 bp)suggested that qBB5.1 was a new locus. The LD heat map drawn following the LD analysis was used to identify candidate genes. The lead SNP of qBB5.1,rs5_361251,was precisely localized to LD block 356,799–369,608 bp, which included the full LOC_Os05g01610 sequence (Fig. 4A). Using SMART to predict specific protein domains, we determined that LOC_Os05g01610 encodes a classic PRAF protein comprising RCC1, FYVE, and BRX domains (Fig. 5B).Thus, it was designated as OsPRAF2. Haplotype analyses were performed using six nonsynonymous SNPs in the coding region and five haplotypes shared by at least 10 accessions were detected(Fig. 4B). There were no significant differences in the LL among the accessions with Hap1, Hap2, Hap3, or Hap4 and between the accessions with Hap3 and those with Hap5 after the inoculation with C5 (Fig. 4C). Following the inoculation with IV, the LL varied among the accessions carrying different haplotypes, but the average LL was not less than 5 cm regardless of the haplotype(Fig.4D).These findings suggested that OsPRAF2 was likely associated with the resistance to C5 and IV.
We detected the LOC_Os06g03770 (OsATM3), which encodes a putative ABC transporter/ATP-binding protein in the local LD region of qBB6,which was associated with C5 resistance in the whole population. This gene was located 259 bp from the lead SNP(rs6_1502856).On the basis of the nonsynonymous SNPs,four major haplotypes were distinguished in 383 accessions (Fig. S5A). The accessions with Hap3 had shorter lesions than the accessions carrying Hap1 or Hap2.There were no LL differences between the accessions carrying Hap4 and those carrying Hap1 or Hap3(Fig.S5B).

Fig. 4. Associated region qBB5.1 for lesion length and haplotype analysis of OsPRAF2. (A) Local Manhattan plot (top) and linkage disequilibrium heat map (bottom) for the region surrounding the lead SNP.(B)Gene structure and haplotype analyses of OsPRAF2.Lesion lengths of accessions with different haplotypes inoculated with C5(C)and IV(D). Different characters above boxplots indicate significant differences according to Duncan’s multiple comparisons test (P <0.05).
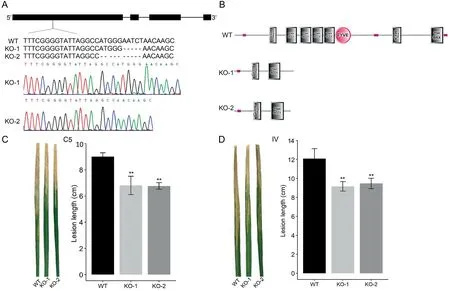
Fig. 5. Functional characterization of OsPRAF2 based on a CRISPR-Cpf1 gene editing strategy. (A) Mutations in the two knockout lines in the Nipponbare background. (B)Predicted proteins in the two mutants and wild-type plants.Inoculation of the knockout lines and wild-type plants with C5(C)and IV(D).WT,wild-type Nipponbare;KO-1 and KO-2, two independent knockout lines; Error bars represent the mean ± SD (n ≥30); **, P <0.01.
The QTL qBB4,qBB8,qBB9,qBB11.1, and qBB11.3 were exclusive to the XI subgroup and were involved in the resistance to IV.More specifically, qBB4 was localized to 21,849,622–21,855,358 bp on chromosome 4, which included one annotated gene(LOC_Os04g35860). This gene encodes a T-complex 11 family protein, similar to the Arabidopsis thaliana orthologs AT1G22930 and AT4G09150 involved in signal transduction. In contrast, qBB8 was detected in a 162-kb region(3,850,174–4,012,319 bp) on chromosome 8.This region harbored a terpene synthase(TPS)gene cluster,including LOC_Os08g07080 (OsTPS30), LOC_Os08g07100 (OsTPS31),and LOC_Os08g07120, which influence rice phytoalexin synthesis.Additionally, qBB11.1 contained an F-box gene cluster comprising LOC_Os11g09418 (OsFBX399), LOC_Os11g09474 (OsFBX400),LOC_Os11g09478 (OsFBX402), and LOC_Os11g09510 (OsFBX403),which were predicted to contribute to biotic stress resistance.We mapped qBB11.3 to a 108-kb region(17,397,638–17,506,154 b p) on chromosome 11. In this genomic interval, we detected missense mutations in LOC_Os11g30060, encoding a typical nucleotide-binding and leucine-rich repeat (NLR) protein. Thus,LOC_Os11g30060 was revealed as a candidate gene for qBB11.3.The qBB9 region(7,820,737–7,853,206 bp)contained four putative annotated genes (Fig. S6A). Of these genes, LOC_Os09g13510 and LOC_Os09g13540 were annotated as retrotransposons, whereas LOC_Os09g13520 encodes a hypothetical protein and LOC_Os09g13530 (OsC3H58) encodes a zinc finger C-x8-C-x5-Cx3-H-type protein. Using nonsynonymous SNPs (rs9_7843377,rs9_7843500, and rs9_7844099), we identified two major haplotypes, with Hap1 accounting for 84% of the accessions in the XI subgroup(Fig.S6B).There were significant LL differences between the two haplotypes,and Hap1 of OsC3H58 conferred a higher level of resistance to IV(Fig.S6C).These results suggested OsC3H58 may affect BB resistance,but this possibility will need to be experimentally verified.
The qBB11.2 QTL influencing C5 resistance in the whole population was delimited to a 378-kb region(16,390,794–16,769,495 bp)on chromosome 11. In the LD block, the gene adjacent to the lead SNP was LOC_Os11g28920, which encodes an expressed protein.In contrast, qBB11.4 was associated with IV resistance in the whole population and the XI population. The qBB11.4 genomic region included the receptor-like kinase gene LOC_Os11g45540(OsRLCK352),as well as the stress resistance gene LOC_Os11g45620,which was identified as a homolog of the leaf rust resistance gene Lr21. Four major haplotypes of LOC_Os11g45540 were detected(Fig. S7A), and accessions with Hap3 were the most resistant to BB(Fig.S7B).Two major haplotypes of LOC_Os11g45620 were identified (Fig. S7C), and accessions with Hap2 had shorter lesions(Fig. S7D). We determined that qBB11.5 and qBB11.6 were associated with C5 and IV resistance in the whole population and were involved in IV resistance in the XI population. A typical NLR resistance gene,LOC_Os11g46210(Pik),was localized in the LD block of qBB11.5, which implies that it may be a suitable candidate gene.The qBB11.6 genomic interval contained some disease resistancerelated genes, including a receptor kinase gene(LOC_Os11g47290),an NLR gene(LOC_Os11g47447),a C2H2-type zinc finger transcription factor gene(LOC_Os11g47620),and two Class III chitinase gene homologs LOC_Os11g47500 (OsChib3H-b) and LOC_Os11g47600(OsChib3H-h).
3.5. OsPRAF2 negatively regulates BB resistance
To verify that the protein encoded by the candidate gene for qBB5.1(OsPRAF2)affects rice BB resistance,we generated knockout(KO) mutants in the japonica (geng) cultivar Nipponbare background using a CRISPR-Cpf1 approach.Two independent OsPRAF2-KO lines, in which a short fragment (10 or 5 bp) was deleted from the target site, were identified among the T2transgenic plants on the basis of Sanger sequencing results(Fig.5A).A premature translation termination in both KO lines resulted in the production of truncated proteins (Fig. 5B). After the inoculation with Xoo strain C5, the KO plants had shorter leaf lesions (6.1–6.5 cm) than the wild-type plants (9.2 cm). Similarly, the leaf lesions on the KO plants were approximately 25% shorter than the leaf lesions on the wild-type plants following the inoculation with IV (Fig. 5C,D).These observations suggested that OsPRAF2 may negatively regulate rice resistance to BB. Moreover, genetic modifications introduced via CRISPR-Cpf1 may be useful for breeding new rice cultivars exhibiting increased disease resistance.
4. Discussion
Bacterial blight is one of the most devastating diseases of rice,causing significant yield losses throughout Asia [12]. There is currently no effective method for managing BB because of the environmental pollution associated with pesticide applications and the rapid development of virulent Xoo strains[61].In South China,where rice BB outbreaks can be severe, new virulent Xoo strains were identified capable of overcoming the resistance conferred by Xa4 and Xa21[31].Therefore,there is an urgent need for screening disease-resistant cultivars to identify key functional genes applicable for breeding new varieties with enhanced BB resistance.In a previous study, 172 indica rice germplasm were tested for BB resistance, and only eight and four accessions were moderately resistant (5 cm ≤LL <10 cm) to the prevalent Chinese strains C5 and IV, respectively [42]. In the present study, we identified 48 accessions that are highly resistant to both C5 and IV, with more than 70% of the accessions originating from Bangladesh(Table S3). The data generated in this study regarding these BBresistant accessions may be relevant for rice breeding programs.
4.1.Analyses of the genetic loci identified in this study with known BB resistance-related genes and QTL
In the past few decades, many investigations on rice BB resistance focused on identifying genetic loci through linkage mapping or association analyses [11,14,16,17,42,61–63]. In the current study, we detected a new locus qBB9, which is related to IV resistance,far from L8,L9, and QBbr9 previously identified in two populations [37,42]. We also identified genetic loci that overlapped with cloned or fine-mapped genes (Table 1). For example, qBB5.2 overlapped with xa5 [17]; qBB11.5 overlapped with Xa35(t), Xa36(t), Xa40, Xa43(t), and xa44(t) [11,12,61–63]; and qBB11.6 overlapped with Xa35(t) and Xa3/Xa26 [16,62]. Surprisingly, Xa35(t)spanned a genomic region overlapping with three QTL (qBB11.4,qBB11.5, and qBB11.6) on chromosome 11, which suggests that high-resolution SNPs can narrow down fine-mapped genes and facilitate gene cloning and functional characterization.
Because of the availability of large-scale sequencing data,GWAS has become a popular and powerful approach for detecting variants associated with a particular trait [32]. Regarding BB, several studies involving natural populations revealed loci associated with resistance to various Xoo strains [35,37,42,53,64]. Some identified loci were enriched on chromosomes 5 and 11, including loci detected in this study (Fig. 3). In contrast to previously identified QTL, some QTL detected in the current study may confer broad spectrum resistance to BB.For example,qBB5.2, which contributes to the resistance to C5 and IV, was located in the same region as qBLB5.1, which confers resistance to PXO86 [36]. Additionally,qBB6 overlapped with QBbr6, which was mapped in a MAGIC population derived from eight parents [37]. Both qBB11.4 and qBB11.5 overlapped with Xa35(t), Xa36(t), L11 conferring resistance to P1 and P6, and qBLB11.1 conferring resistance to PXO61, PXO99, and PXO86 [36,42,62,63]. Moreover, qBB11.4 overlapped with QBbr11-1,whereas qBB11.5 and qBB11.6 overlapped with QBbr11-2 [37]. The remaining three QTL on chromosome 11 (qBB11.1, qBB11.2, and qBB11.3) as well as qBB1, qBB4, qBB5.1, qBB8, and qBB9 were not associated with previously reported genes/QTL in biparental and association populations, which implies that they may be new genetic loci mediating BB resistance. These findings provide rice researchers and breeders with new information that may be useful for improving BB resistance.
4.2. Resistance genes underlying QTL
In plants,many NLR proteins function together to induce immunity [65]. In this study, we identified some NLR genes in QTL regions, including LOC_Os01g06920 within qBB1, LOC_Os11g30060 within qBB11.3, LOC_Os11g45620 within qBB11.4, LOC_Os11g46210(Pik) within qBB11.5, and LOC_Os11g47447 within qBB11.6.Receptor-like kinases are often considered as pattern recognition receptors that influence plant immunity [66]. Our analyses detected the receptor-like kinase genes LOC_Os11g45540(OsRLCK352) and LOC_Os11g47290 in the candidate regions of qBB11.4 and qBB11.6, respectively. Zinc finger proteins, which are common transcription factors in rice, have crucial functions in a series of regulatory processes [67]. An earlier investigation indicated that a natural allele of a transcription factor gene, bsr-d1,confers broad-spectrum blast resistance [68]. Similar to bsr-d1, a C2H2-type zinc finger transcription factor gene (LOC_Os11g47620)within qBB11.6 may contribute to IV resistance. Moreover, haplotype analyses of the zinc finger C-x8-C-x5-C-x3-H-type gene OsC3H58 associated with qBB9 indicated that accessions with Hap1 have shorter lesions than accessions with Hap2 (Fig. S6),which suggests that OsC3H58 is a candidate disease resistancerelated gene.
Notably,the following three gene clusters involved in BB resistance were identified: TPS gene cluster, F-box gene cluster, and chitinase gene cluster. A diterpenoid gene cluster on chromosome 7 (DGC7) comprising OsTPS28, OsCYP71Z2, and OsCYP71Z2 was reported to synthesize 5,10-diketo-casbene,which is a monocyclic casbene-derived diterpenoid that functions as an antimicrobial phytoalexin that mediates rice immunity against Magnaporthe oryzae and Xoo [69]. Similar to DGC7, three TPS genes (OsTPS30,OsTPS31,and LOC_Os08g07120)were detected in the qBB8 genomic interval,which suggests that these genes may confer BB resistance.Several F-box proteins have been implicated in plant defense responses. The overexpression of the F-box gene OsDRF1 in transgenic tobacco upregulates the expression of defense-related genes,thereby increasing plant resistance to Pseudomonas syringae pv.tabaci and tomato mosaic virus [70,71]. Although OsFBX399,OsFBX400,OsFBX402,and OsFBX403 were detected in the candidate region of qBB11.1, their functions remain unclear. Plant chitinases usually catalyze the breakdown of chitin in the cell wall of pathogens and the release of defense-related substances in response to various biotic and abiotic stresses [72]. In sweet potato, a new Class II extracellular chitinase encoded by IbChiA has remarkable fungicidal effects on Ceratocystis fimbriata[73].In rice, the expression of the chitinase gene LOC_Os04g41620 and the defense-related gene OsPR1a is directly regulated by OsWRKY114 and increases plant resistance to Xoo[74].In the present study,two Class III chitinase genes, OsChib3H-b and OsChib3H-h, were detected within qBB11.6. These gene clusters may provide clues regarding the mechanism underlying BB resistance in rice.
The plant-specific PRAF protein family members contain the following distinct domains: PH, RCC21, FYVE, and BRX [75]. Their functions in plant development have rarely been reported.Arabidopsis thaliana contains nine PRAF proteins with similar structures, but mutations in these PRAF proteins do not result in obvious phenotypic changes [76]. In Medicago truncatula, the PRAF protein MtZR1 is reportedly involved in the development of roots and symbiotic root nodules, thereby affecting nitrogen fixation efficiency[77].In rice,there are five genes encoding PRAF proteins,but they will need to be functionally characterized in future investigations.
In the present study, we proved that knocking out OsPRAF2 within qBB5.1 significantly enhances plant resistance to C5 and IV, which implies that OsPRAF2 negatively regulates BB resistance.Although the molecular mechanism underlying PRAF-mediated disease resistance is unclear, our results indicate that editing OsPRAF2 using the CRISPR system may be a viable approach for enhancing the resistance of rice cultivars to BB.
5. Conclusions
A panel of 421 diverse rice accessions and 3,080,542 SNPs were used to perform a GWAS to detect QTL associated with the resistance to Xoo pathotypes prevalent in South China. A total of 48 accessions were highly resistant to the virulent strains C5 and IV.Additionally, 13 loci were associated with BB resistance, including five known and eight new QTL.Editing OsPRAF2 within the new loci qBB5.1 resulted in increased resistance to C5 and IV.The identified QTL and genes are potentially useful for improving rice BB resistance in South China.
CRediT authorship contribution statement
Jialing Lu:Investigation,Data curation,Writing-original draft.Quanlin Li:Investigation,Data curation.Chunchao Wang:Investigation, Data curation.Mingming Wang:Investigation, Data curation.Dan Zeng:Investigation, Data curation.Fan Zhang:Investigation, Data curation.Wenxue Zhai:Supervision, Writing- review & editing.Yongli Zhou:Project administration, Supervision, Writing - review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We thank Prof. Liexian Zeng (Guangdong Academy of Agricultural Sciences) for providing Xoo strain IV and Jiankang Zhu(Shanghai Center for Plant Stress Biology, Chinese Academy of Sciences) for providing the CRISPR-Cpf1 system. This research was supported by the National Natural Science Foundation of China (31661143009 and 31571632), the CAAS Innovative Team Award, and the Bill & Melinda Gates Foundation (OPP51587). The funders were not involved in designing the study, collecting, analyzing, and interpreting the data, or writing the manuscript.
Appendix A. Supplementary data
Supplementary data for this article can be found online at https://doi.org/10.1016/j.cj.2021.05.009.

- The Crop Journal的其它文章
- Origin, evolution, and molecular function of DELLA proteins in plants
- Far-red light: A regulator of plant morphology and photosynthetic capacity
- A rice XANTHINE DEHYDROGENASE gene regulates leaf senescence and response to abiotic stresses
- Dissection of heterotic loci for grain yield using interconnected chromosome segment substitution lines in rice
- A soybean NAC homolog contributes to resistance to Phytophthora sojae mediated by dirigent proteins
- The boron transporter SiBOR1 functions in cell wall integrity, cellular homeostasis, and panicle development in foxtail millet