
钠牛磺胆酸共转运多肽缺陷病临床特征及SLC10A1基因突变分析
2022-03-28 07:30杨峰霞曾凡森谭丽梅刘玲丽
临床肝胆病杂志 2022年3期
杨峰霞,曾凡森,谭丽梅,龚 余,刘玲丽,徐 翼
广州市妇女儿童医疗中心 感染性疾病科,广州 510120
钠牛磺胆酸共转运多肽(sodium taurocholate cotrans-porting polypeptide,NTCP)缺陷病是由于溶质转运蛋白家族10成员1(solute carrier family 10 member 1,SLC10A1)基因突变引起的一种遗传性胆汁酸代谢病。NTCP是一种表达于肝细胞基侧膜的转运蛋白,由定位于染色体14q24.2 的基因 SLC10A1 编码,其主要功能是以钠依赖方式,将结合型胆汁酸从血浆摄取入肝细胞,在胆汁酸肠肝循环中发挥重要作用。因此,NTCP的缺乏将导致血液循环中胆汁酸水平升高[1]。胆汁酸是胆汁的重要成分,主要存在于肠肝循环中。大约95%的胆汁酸被肝脏重新吸收并循环利用[2]。在正常生理条件下,胆汁酸维持恒定的内部环境,并具有抗炎特性。然而,胆汁淤积会导致肝脏炎症和损伤。因此,维持血液循环和肝脏中相对较低的胆汁酸水平是非常重要的[3]。此外,某些甾体激素、甲状腺激素、药物以及药物与胆汁酸的结合体也是NTCP摄取的底物[4]。近年研究[5-6]发现,NTCP还是HBV和HDV进入肝细胞的受体。荷兰学者Vaz等[7]于2015年报道了全球首例NTCP缺陷病。截至目前,医学界对NTCP缺陷病的认识仍主要基于散发性的病例报道。作为一种新的罕见的遗传性胆汁酸代谢病,NTCP缺陷病的临床表型、生化特征和SLC10A1基因型特征尚需进一步研究总结,以促进临床早期识别,避免不必要的诊疗干预和人群焦虑。本研究基于本中心经基因检测确诊的 10 例NTCP缺陷病患者,对该病临床表现、生化特点及基因突变进行分析总结。
1 资料与方法
1.1 研究对象 选取2020年6月—2021年6月于本中心因持续性高胆汁酸血症就诊且基因检测具有SLC10A1基因纯合或复合杂合突变的10例18岁以下患儿为研究对象。排除感染性、结构性和免疫性胆汁淤积患儿。
1.2 研究方法 收集10例患儿的一般资料(性别、年龄、身高、体质量、家族史和既往病史)、临床表现、病情转归、实验室检查(血常规、肝功能、嗜肝病毒、自身免疫性肝炎筛查)及基因突变检测结果,进行回顾性分析总结。基因检测及家系验证流程:家属签署知情同意书后,抽取患儿及其父母外周静脉血2.0 mL置于EDTA抗凝管中,混匀后即送至武汉康圣环球有限公司进行基因测序。采用安捷伦外显子芯片捕获+高通量测序法对先证者进行60个与胆汁淤积性黄疸相关基因的检测,检测到突变基因后再用sanger测序法对患儿及其父母基因突变进行验证,将测序发现的突变在人类基因突变数据库(GMD,www.hgmd.org)、人类孟德尔遗传数据库(OMIM,www.ncbi.nlm.nih.gov/omim)对基因对应的疾病及遗传方式、HGMDpro数据库收录情况进行比对,按照美国医学遗传学和基因组学会(ACMG)遗传变异分类标准指南[8]对新突变分级。
2 结果
2.1 一般资料 10例患儿均来自不同的家庭,按就诊时间排序编号P1~P10,其中男8例,女2例,男女比例为4∶1。身高/体质量均在同龄同性别儿童的25%~75%之间(参照2009年中国0~3岁和2~18岁儿童身高/体质量百分位曲线);确诊年龄3~37个月,中位年龄11.5个月;P2患儿35周早产,P6患儿足月出生,但出生体质量<2.5 kg(足月小样儿),余患儿出生史无异常。P4患儿母亲孕期合并妊娠期肝内胆汁淤积症。首次就诊病因包括新生儿黄疸延长(5/10,50%)、转氨酶水平升高(2/10,20%)、体检(2/10,20%)和肺炎(1/10,10%)。肝脾肿大患儿1例(P6),肝右肋下3 cm,脾肋下3 cm,质地中等。所有患儿均无皮肤瘙痒及黄瘤(表1)。
2.2 实验室检查结果 所有患儿血清TBA水平均升高,平均水平(154.8±90.1 μmol/L);ALT、AST水平升高2例;TBil水平升高1例,且以DBil水平升高为主(DBil/TBil>50%);P6患儿住院期间检测巨细胞病毒抗体IgM弱阳性,血浆巨细胞病毒DNA 601拷贝/mL,脑干听觉诱发电位、胸部X线检查未见异常,病程中曾予更昔洛韦注射液抗病毒治疗2周,但疗效欠佳,TBil、DBil水平较治疗前无明显下降,由此推断因巨细胞病毒感染引起胆汁淤积的可能性较小;其他患儿HAV、HBV、HCV、HEV、EB病毒、巨细胞病毒、微小病毒B19、自身免疫性肝炎、梅毒、HIV检测及尿有机酸分析均未见异常(表1)。
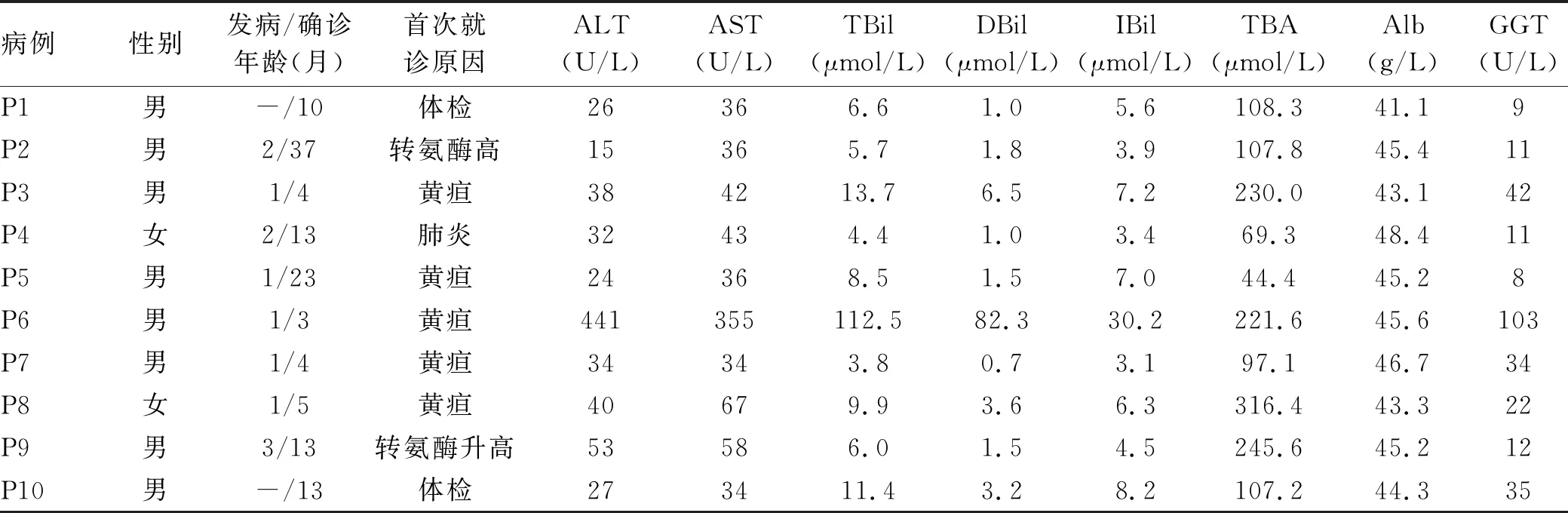
表1 确诊时患儿主要临床表现、实验室检查结果
2.3 基因检测结果 经第二代基因测序,10例患儿均为SLC10A1基因纯合突变:c.800C>T(p.Ser267Phe,chr14∶70245193)。父亲均为杂合突变,P1、P4、P10患儿母亲为纯合突变。其中,P4患儿母亲妊娠期间有肝内胆汁淤积症,P1、P10患儿母亲无症状,但血TBA水平(18.4 μmol/L、21.6 μmol/L)稍高于正常值上限。其他胆汁淤积相关基因:P1、P8、P10患儿为UGT1AI杂合突变,P5患儿为UGT1A1纯合突变、ABCB4杂合突变,P6患儿为UGT1A1复合杂合突变,P7患儿为TJP2杂合突变(表2)。
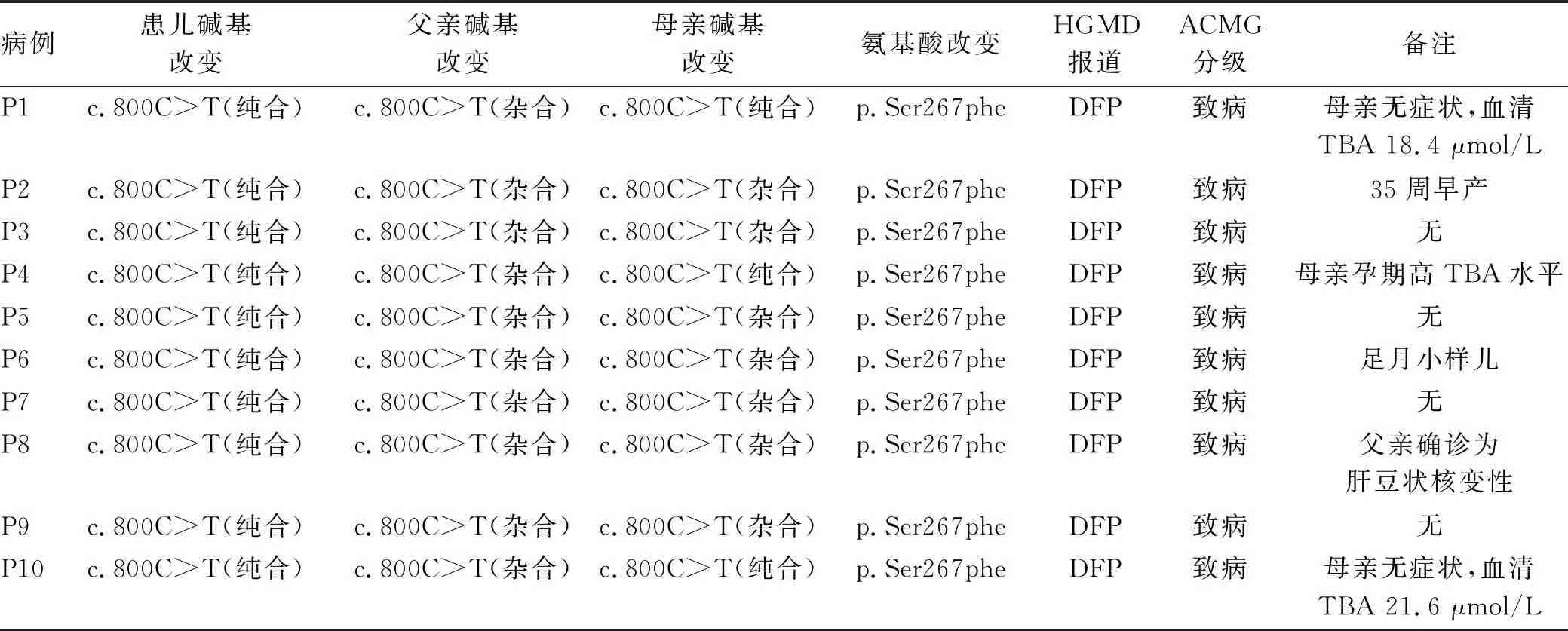
表2 10例患儿及其父母第二代基因检测结果
3 讨论
NTCP缺陷病是近年来逐渐被认识的一种新的遗传性胆汁酸代谢病,属于常染色体隐性遗传病[9]。NTCP缺陷病是由于SLC10A1基因突变,影响NTCP从血浆中摄取胆汁酸盐的功能,导致胆汁酸在血液中大量堆积,形成显著而顽固的高胆汁酸血症。由于胆汁酸的合成、分泌、重吸收等均未受到直接影响,因此NTCP缺陷患者除了高胆汁酸血症,其他临床表现可不明显[1],这有别于引起胆汁酸水平增高的其他遗传代谢性疾病。本研究中10例NTCP缺陷病患儿生长发育均正常,诊断时年龄最小3个月,最大37个月。受影响的男孩多于女孩,第1次就诊的原因不尽相同,最常见的是黄疸。除高胆汁酸血症外,最常见的生化特征为ALT、AST、TBil水平升高(年幼婴儿多见),随着治疗及年龄增长,转氨酶和胆红素指标逐渐恢复正常,但无法纠正高胆汁酸血症,与其他肝功能指标变化趋势不一致,这是NTCP缺陷病的特征性表现。NTCP缺陷病早期可能表现为短暂的婴儿胆汁淤积症,使临床诊断时错误考虑为胆道闭锁或其他胆管异常,导致过渡干预,如内镜逆行胰胆管造影和胆管探查;后期则仅表现为血胆汁酸水平升高,无黄疸、生长发育迟缓、黄瘤、瘙痒等临床表现,易漏诊。因此,常规将TBA纳入肝功能检查可减少漏诊率。
笔者团队检索国内外文献数据库2015年1月—2021年1月共析出13项研究[7,10-21],涉及74例NTCP缺陷病患儿,除1例来自阿富汗,其他均来自我国。其中,4例存在生长、发育迟缓;首次就诊的原因包括高胆红素血症、新生儿黄疸延长、新生儿短暂胆汁淤积及体检、志愿者招募、肺炎、早产等非肝脏相关疾病;突变位点包括c.800C>T、c.776G>A、c.595A>C、c.755G>a、c.615-618del、c.682-683del、c.263T>c。在73例我国患儿中,c.800C>T纯合突变的携带率为90%(66/73),可视为我国NTCP缺陷病患儿的热点突变。本研究中10例患儿均为c.800C>T (p.Ser267Phe)纯合突变,ACMG分级均为致病,其中P1、P4、P10患儿母亲也为c.800C>T纯合突变,临床表现为轻度高胆汁酸血症及妊娠胆汁淤积症。6例患儿的突变与其他影响胆红素代谢的基因突变(包括UGT1A1、ABCB4、TJP2)合并。根据遗传方式,P5、P6患儿(UGT1A1纯合/复合杂合突变)符合常染色体隐性遗传规律,但与患儿临床表现不相符合,UGT1A1基因突变临床表现为高间接胆红素血症;ABCB4、TJP2为单杂合突变,均与进行性家族性胆汁淤积症相关,但是否会影响胆红素的代谢尚需进一步开展分子水平的研究。
NTCP缺陷病尚无特异性治疗手段,对症支持治疗是主要的管理手段,一般不需要创伤性检查或治疗。患儿短期临床结局均良好,迄今未见有因NTCP缺陷病致死亡或肝硬化等严重预后的报道[22]。然而,高浓度血浆TBA水平对人体健康有何影响,仍需长期随访观察。
综上所述,儿童NTCP患者就诊的最常见原因是黄疸,也可发现于体检或其他非肝脏疾病检查中。黄疸多是暂时的,消退后可无明显临床表现,显著而持续性的高胆汁酸血症提示本病可能。SLC10A1基因分析发现双等位基因致病性变异是可靠的确诊依据,我国的热点突变是c.800C>T (p.Ser267Phe)。因此,对伴有显著而持续性高胆汁酸血症的新生儿高胆红素血症、婴儿早期胆汁淤积症和妊娠胆汁淤积症患者,均有必要行SLC10A1基因分析,以排除NTCP缺陷病可能。
伦理学声明:本研究方案于2019年8月22日经由广州市妇女儿童医疗中心伦理委员会批准,批号:穗妇儿2019-32400,所有检查均取得患儿家属知情同意。
利益冲突声明:本研究不存在研究者、伦理委员会成员、受试者监护人以及与公开研究成果有关的利益冲突。
作者贡献声明:杨峰霞负责课题设计,资料分析,撰写论文;曾凡森、谭丽梅、龚余、刘玲丽参与收集数据,修改论文;徐翼负责拟定写作思路,指导撰写文章并最后定稿。
猜你喜欢
珠江水运(2022年17期)2022-09-25
珠江水运(2021年15期)2021-08-29
家庭医学·下半月(2021年1期)2021-03-28
科学与财富(2020年33期)2020-03-10
家庭医学(2018年2期)2018-07-14
考试周刊(2017年26期)2017-12-12
黄河黄土黄种人(2017年11期)2017-11-27
水能经济(2017年6期)2017-10-19
家庭百事通·健康一点通(2017年8期)2017-08-18
校园英语·下旬(2017年7期)2017-07-14
