
适用于线粒体基因组测序的高纯度线粒体DNA提取和鉴定方法优化
2022-03-27 10:32李君朱嘉晋邓雨萍吴文鹤
中国现代医生 2022年4期
李君 朱嘉晋 邓雨萍 吴文鹤



[摘要] 目的 优化线粒体DNA(mtDNA)提取和纯度鉴定方法以满足线粒体基因组测序的要求。方法 利用简化的蔗糖密度梯度离心法、DNA酶Ⅰ并结合质粒提取试剂盒提取纯化mtDNA、尽量降低核DNA(nDNA)污染的可能。获得的mtDNA经NanoDrop检测、琼脂糖凝胶电泳和荧光定量PCR绝对定量法测定nDNA/mtDNA拷贝数比值等鉴定其纯度。结果 从小鼠肝脏和大脑组织中提取纯化得到mtDNA产率分别为311.7 μg/g和59.1 μg/g,其A260/280均介于1.8~2.0,A260/230浓度均大于2.0,荧光定量PCR绝对定量法测定nDNA/mtDNA拷贝数比值均低于0.005%。结论 本研究优化了mtDNA提取和鉴定方法,得到的高纯度mtDNA适用于后续线粒体基因组测序等分子生物学研究。
[关键词] 线粒体DNA;提取;纯化;核DNA
[中图分类号] R394.3 [文献标识码] A [文章编号] 1673-9701(2022)04-0040-04
[Abstract] Objective To optimize mitochondrial DNA (mtDNA) extraction and purity identification methods to meet the requirements of mitochondrial genome sequencing. Methods Simplified sucrose density gradient centrifugation, DNase I, and plasmid extraction kit were used to extract and purify mtDNA and minimize the possibility of nuclear DNA (nDNA) contamination. The purity of the obtained mtDNA was determined by NanoDrop detection, agarose gel electrophoresis, and determination of nDNA/mtDNA copy number ratio by fluorescence quantitative PCR absolute quantitative method. Results The yields of mtDNA extracted and purified from mouse liver and brain tissues were 311.7 μg/g and 59.1 μg/g, respectively. The A260/280 ranged from 1.8 to 2.0, and the A260/230 concentration was greater than that of 2.0. The ratio of nDNA/mtDNA copy number determined by fluorescence quantitative PCR absolute quantitative method was lower than 0.005%. Conclusion This study optimizes the mtDNA extraction and identification methods, and the high purity mtDNA obtained is suitable for subsequent mitochondrial genome sequencing and other molecular biology research.
[Key words] Mitochondrial DNA; Extraction; Purification; Nuclear DNA
隨着测序技术的快速蓬勃发展,获取线粒体基因组序列变得更为容易[1],如何高效分离和纯化线粒体DNA(mitochondrial DNA,mtDNA)而避免核DNA(nuclear DNA,nDNA)的污染是线粒体基因组测序及后续序列分析的关键[2]。另外,现有的DNA纯度鉴定方法多采用PCR扩增后通过琼脂糖凝胶电泳鉴定,其敏感度较低且操作较繁琐。因此,本研究通过对现有mtDNA提取和鉴定方法进行优化,获得高纯度的mtDNA,以满足线粒体基因组测序和后续相关分子生物学研究的要求。
1 材料与方法
1.1 材料来源
本研究经学校动物伦理委员会批准,选取新鲜C57BL/6小鼠[动物许可证号SYXK(浙)2015-0009]肝脏和大脑组织200 mg进行后续实验。
1.2 仪器与试剂
CFX Connect实时定量PCR仪(BIO-RAD,美国),NanoDrop one(Thermo Scientific,美国)。DNA酶Ⅰ(deoxyribonuclease Ⅰ,DNase Ⅰ)(NEB,美国),质粒DNA小提试剂盒(Qiagen,美国),BCA蛋白定量分析试剂盒(Pierce,美国)。H缓冲液(10 mM Tris-HCl(pH 7.4)/250 mM 蔗糖/1 mM EDTA缓冲液),W缓冲液[10 mM Tris-HCl(pH 7.4)/250 mM 蔗糖],S缓冲液(洗涤缓冲液/30%蔗糖),均为国产分析纯。
1.3 方法
1.3.1 线粒体粗提 将新鲜小鼠大脑组织切成小块(约1 mm3),4℃、500×g离心3 min,取沉淀加入5倍体积预冷的H缓冲液,转移至预冷的玻璃匀浆器(高压灭菌)中进行匀浆,然后在4℃、1000×g离心10 min,上清即为粗提线粒体。
1.3.2 线粒体纯化 利用简化的蔗糖密度梯度离心进一步纯化线粒体[3-4]。将上述粗提线粒体,4℃、15 000×g离心15 min,取沉淀用1 ml预冷的H缓冲液重悬,轻轻加到30%蔗糖缓冲液上,在4℃、2400×g水平离心5 min;离心后,小心吸取3个分层中最上面一层悬浮液并转入新离心管;加5倍体积预冷W缓冲液重悬,4℃、15 000×g离心10 min,纯化后的线粒体处于离心管壁上的少量沉淀中。
1.3.3 mtDNA提取和浓度测定 利用DNA酶去除nDNA污染,进一步纯化线粒体[5]。用500 μl W缓冲液重悬,加入DNase Ⅰ和MgCl2(终浓度分别为10 U/ml和2.5 mM),混匀,冰上孵育1 h。加入pH 8.0 EDTA至终浓度50 mM终止DNase Ⅰ酶切反应。4℃、15 000×g离心10 min,沉淀用质粒DNA小提试剂盒提取mtDNA。采用NanoDrop测定DNA浓度和纯度初测,有无蛋白污染用BCA蛋白定量分析试剂盒测定。
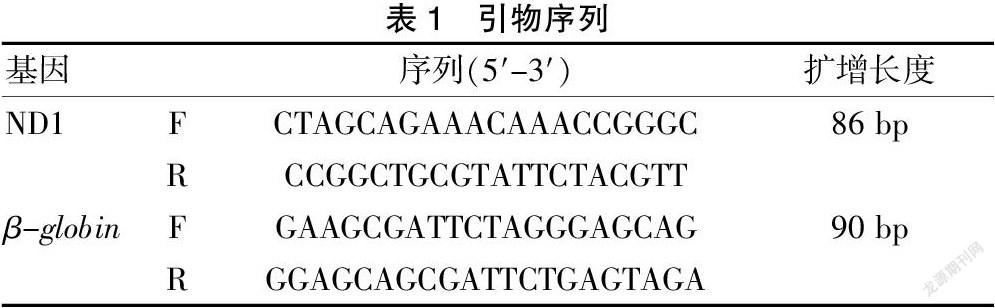
1.3.4 荧光定量PCR绝对定量法测定mtDNA和nDNA含量 构建分别包含小鼠线粒体基因和核基因的两组标准质粒,线粒体基因选用ND1基因[6-8],核基因选用β-globin基因[9-10],通过普通PCR扩增目的片段,T-A克隆,构建pCRTM2.1-TOPO-ND1(526 bp)和pCRTM 2.1-TOPO-globin(551 bp)重组质粒,菌落PCR鉴定克隆并选取阳性克隆测序确认载体构建成功。构建好的质粒经测序鉴定无误后用Thermo Nanodrop测定质粒A260的值,通过公式换算成拷贝数(copies/μl),计算公式如下:质粒浓度换算公式(copies/μl)=浓度(ng/μl)×6.02×1014/分子量。将构建好的标准质粒依次十倍稀释成108~10 copies/μl标准曲线样本,并与分析样本同时进行荧光定量PCR分析[11-12],同一样本分别扩增ND1和β-globin,所用引物见表1。实时定量PCR反应体系为20 μl:SYBR Green master mix(2×)10 μl,10 μM上下游引物各1 μl,模板5 μl,H2O 3 μl。PCR反应条件为:95℃ 10 min;循环40次:95℃ 15 s,60℃ 1 min。融解曲线:95℃ 15 s,60℃ 1 min,95℃ 15 s。每个样品重复3次,并做阴性对照。mtDNA样本中检测到nDNA拷贝数低于0.005%则可以用于后续的线粒体基因组测序。
2 结果
2.1 mtDNA浓度测定和纯度初测
将纯化后的mtDNA样本经NanoDrop测定分析,结果显示从200 mg的小鼠肝脏和大脑组织中,分别得到62.34 μg和11.82 μg mtDNA,即产量分别为311.7 μg/g和59.1 μg/g;其A260/280均介于1.8~2.0,A260/230浓度均大于2.0,说明样本无蛋白质和无机盐污染。将纯化后的mtDNA样本经琼脂糖凝胶电泳分析,紫外灯下可观察到一条清晰的大小约16 kb的条带,说明样本无降解。见表2。
2.2 建立mtDNA和nDNA拷贝数检测方法
2.2.1 标准质粒测序结果及比对 利用NCBI在线BLAST工具,pCRTM2.1-TOPO-ND1和pCRTM2.1-TOPO-globin两个重组质粒测序结果分别与GeneBank上公布的C57BL/6小鼠线粒体ND1基因和β-globin基因比对,同源性为100%和99%。结合之前的菌落PCR结果,可以确认分别含ND1基因526 bp片段和β-globin基因551 bp片段的两个标准质粒构建成功。其中,pCRTM2.1-TOPO-ND1大小为4457 bp,通过全部序列计算分子量为2753 915.7 g/mol;pCRTM2.1-TOPO-globin大小为4482 bp,通过全部序列计算分子量为2769 389.1 g/mol。
2.2.2 标准曲线建立 如图1(a)所示,ND1标准曲线方程为Y=-3.336X+34.91,其中R2=0.9986,扩增效率=99.42%;如图1(b)所示,β-globin标准曲线方程为Y=-3.343X+35.69,其中R2=0.9995,扩增效率=99.12%。说明ND1和β-globin两个基因的扩增效率基本一致,均接近100%,且重复性好,适于样本的分析。
2.3荧光定量PCR绝对定量法测定mtDNA纯度
將纯化后的mtDNA样本与两个质粒标准样品稀释液一起进行荧光定量PCR,通过上述标准曲线得到纯化后的两个mtDNA样本拷贝数,并计算nDNA/mtDNA拷贝数比值。纯化的小鼠肝脏和大脑mtDNA样本中检测到nDNA拷贝数分别为0.0016%和0.0012%,均低于0.005%,说明其中的nDNA量极低,可用于后续的线粒体基因组测序等。见表3。
3讨论
在基因组时代,线粒体基因组和核基因组一样重要。mtDNA具有高拷贝数、无基因重组、高突变率和母系遗传等特点[13],被广泛应用在生物医学的各个领域[14-15]。目前,线粒体基因组测序基本原理是将样本总DNA或分离纯化后的mtDNA随机打断成片段化的单链DNA文库[16],与测序接头序列连接后再进行高通量测序,最后通过生物信息分析从海量的全基因组数据中获取线粒体基因组序列。
由于mtDNA和nDNA之间存在一定的同源性,很难明确地识别随机打断片段的起源。另外,mtDNA的长度远远小于nDNA,如人类和小鼠mtDNA长度都约为16 kb,而相应的nDNA长度却为mtDNA的成千上万倍[2,17],因此即使是极低拷贝数的nDNA污染也会导致获得的单链DNA文库大多是nDNA来源。所以如何高效分离和纯化mtDNA,避免nDNA的污染是线粒体基因组测序及后期拼装分析的关键。而这一步的关键在于先分离到高纯度的线粒体。经典的线粒体分离方法有密度梯度离心法[18-19]、差速离心法[20-21]等。差速离心法是利用细胞中不同组分具有不同的沉降系数,通过不同的离心力将其分离开来,但分离出的线粒体纯度往往不高。而密度梯度离心法是在离心力固定的情况下,通过有机盐、糖类等介质形成不同的密度区域进行细胞分离。目前使用较多的介质有氯化铯和蔗糖,但往往操作复杂、费时,而且氯化铯价格昂贵,操作具有一定的危险性。所以本研究首先通过30%蔗糖缓冲液简化蔗糖密度梯度离心法,去除粗提线粒体中混杂细胞核纯化线粒体。接着利用DNase Ⅰ去除线粒体提取过程中可能的nDNA污染,进一步纯化线粒体[5],再进行mtDNA 的提取和纯化。目前,mtDNA的提取和纯化主要有碱裂解法、高盐沉淀法、Triton法三种,其中碱裂解法应用最为广泛,如多种质粒提取试剂盒。因此,本研究利用质粒提取试剂盒去除nDNA、蛋白质和RNA等污染再一次纯化mtDNA。
另外,現有的DNA纯度鉴定方法多采用PCR扩增后通过琼脂糖凝胶电泳鉴定,其敏感度远远不能满足后续线粒体基因组测序的要求。因此,本研究除了利用NanoDrop检测、琼脂糖凝胶电泳确定样本浓度和初步鉴定纯度,还分别利用含ND1和β-globin的两个标准质粒建立了一种荧光定量PCR绝对定量法测定mtDNA和nDNA拷贝数,进一步确定提取的mtDNA样本纯度。本研究结果显示,经NanoDrop检测,从小鼠肝脏和大脑组织中提取纯化得到mtDNA产率分别为311.7 μg/g和59.1 μg/g,其A260/280均介于1.8~2.0,A260/230浓度均大于2.0,说明样本未见蛋白质和无机盐污染。将纯化后的mtDNA样本经琼脂糖凝胶电泳分析,紫外灯下可观察到一条清晰的大小约16 kb的条带,说明样本无降解。最后通过这种荧光定量PCR绝对定量法检测纯化后的小鼠肝脏和大脑nDNA/mtDNA拷贝数比值分别为0.0016%和0.0012%,均低于0.005%,说明其中的nDNA量极低,可用于后续的线粒体基因组测序等。
[参考文献]
[1] Goudenege D,Bris C,Hoffmann V,et al. eKLIPse:A sensitive tool for the detection and quantification of mitochondrial DNA deletions from next-generation sequencing data[J].Genetics in Medicined,2019,21(6):1407-1416.
[2] Quiros PM,Goyal A,Jha P,et al.Analysis of mtDNA/nDNA ratio in mice[J].Current Protocols in Mouse Biology,2017,7(1):47-54.
[3] Panda AP,Roy SC,Sakhare DT,et al.Reduced cytochrome oxidase activity and increased protein tyrosine phosphorylation of mitochondria-rich fractions of buffalo(Bubalus bubalis)spermatozoa after a cycle of freezing and thawing[J].Reproduction,Fertility and Development,2019,31(10):1567-1580.
[4] Sharaf A,Fussy Z,Tomcala A,et al.Isolation of plastids and mitochondria from Chromera velia[J].Planta,2019,250(5):1731-1741.
[5] Lang BF,Burger G.Purification of mitochondrial and plastid DNA[J].Nature Protocols,2007,2(3):652-660.
[6] Bindi E,Li B,Zhou H,et al.Mitochondrial DNA:A biomarker of disease severity in necrotizing enterocolitis[J].European Journal of Pediatric Surgery,2020,30(1):85-89.
[7] Hoque SAM,Umehara T,Kawai T,et al.Adverse effect of superoxide-induced mitochondrial damage in granulosa cells on follicular development in mouse ovaries[J].Free Radical Biology and Medicine,2020,163:344-355.
[8] Ibis O,Selcuk AY,Sacks BN,et al.Whole mitochondrial genome of long-clawed mole vole(Prometheomys schaposchnikowi)from Turkey,with its phylogenetic relationships[J].Genomics,2020,112(5):3247-3255.
[9] Liu Y,Shen Q,Zhao X,et al.Cell-free mitochondrial DNA in human follicular fluid:A promising bio-marker of blastocyst developmental potential in women undergoing assisted reproductive technology[J].Reproductive Biology and Endocrinology,2019,17(1):54.
[10] Wu J,Ren J,Liu Q,et al.Effects of changes in the levels of damage-associated molecular patterns following continuous veno-venous hemofiltration therapy on outcomes in acute kidney injury patients with sepsis[J].Frontiers in Immunology,2019,9:3052.
[11] Kavlick MF.Development of a triplex mtDNA qPCR assay to assess quantification,degradation,inhibition,and amplification target copy numbers[J].Mitochondrion,2019, 46:41-50.
[12] Wang Y,Liu Y,Zhao Y,et al.Quantification of circadian rhythm in mitochondrial DNA copy number in whole blood,and identification of factors that influence it[J].Cellular and Molecular Biology(Noisy-le-grand),2020, 66(5):179-184.
[13] Ingman M,Kaessmann H,Paabo S,et al.Mitochondrial genome variation and the origin of modern humans[J].Nature,2000,408(6813):708-713.
[14] Haberman Y,Karns R,Dexheimer PJ,et al.Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response[J].Nature Communications,2019, 10(38):1-13.
[15] Reinhardt K,Dowling DK,Morrow EH. Medicine.Mitochondrial replacement,evolution,and the clinic[J].Science,2013,341(6152):1345-1346.
[16] Weissensteiner H,Forer L,Fuchsberger C,et al. mtDNA-Server:Next-generation sequencing data analysis of human mitochondrial DNA in the cloud[J].Nucleic Acids Research,2016,44(W1):W64-69.
[17] Bosworth CM,Grandhi S,Gould MP,et al.Detection and quantification of mitochondrial DNA deletions from next-generation sequence data[J].BMC Bioinformatics,2017, 18(Suppl 12):407.
[18] Su YX,Huang XM,Huang ZS,et al.STAT3 localizes in mitochondria-associated ER membranes instead of in mitochondria[J].Frontiers in Cell and Developmental Biology,2020,8:274.
[19] Tarasenko TA,Subota IY,Tarasenko VI,et al.Plant mitochondrial subfractions have different ability to import DNA[J].Theoretical and Experimental Plant Physiology,2020,32(1):5-18.
[20] Long NP,Min JE,Anh NH,et al.Isolation and metabolic assessment of cancer cell mitochondria[J].Acs Omega,2020,5(42):27 304-27 313.
[21] Fisher KE,Bradbury SP,Coates BS.Prediction of mitochondrial genome-wide variation through sequencing of mitochondrion-enriched extracts[J].Scientific Reports,2020,10(1):19 123.
(收稿日期:2021-02-09)
猜你喜欢
湖北农业科学(2016年20期)2017-02-15
企业技术开发·下旬刊(2016年9期)2016-11-23
中学生物学(2016年10期)2016-11-19
文艺生活·中旬刊(2016年9期)2016-11-07
科技视界(2016年22期)2016-10-18
上海医药(2016年3期)2016-03-23
天津农业科学(2015年9期)2015-09-02
科技视界(2015年25期)2015-09-01
