
Al/Fe2O3纳米铝热剂界面结构和稳定性的周期性密度泛函理论研究
2022-03-17 07:28:02王桂香贡雪东
含能材料 2022年3期
薛 闯,高 贫,王桂香,贡雪东
(1. 南京理工大学化学与化工学院,江苏 南京 210094;2. 国家民用爆破器材质量监督检验中心,江苏 南京 210094)
1 引言
由金属Al 和其他金属氧化物构成的铝热剂能量密度高,在燃烧时能释放大量热而产生很高温度,在冶金、爆破、新材料制备等领域有较多应用[1-3]。然而传统铝热剂中的燃料和氧化剂颗粒较大,接触不紧密,导致其燃速慢,一定程度上影响了铝热剂的更广泛应用。研究表明,将铝热剂的粒度从微米级超细化到纳米级时,反应速率可提高超千倍,因此纳米铝热剂成为当前一个研究热点[4-6]。近年来有许多纳米铝热剂体系被研究报道,其中Al/Fe2O3纳米铝热体系因其高能量密度而备受关注,成为研究最广的铝热剂之一[7-10]。
界面是纳米复合材料的重要组成部分,是影响载荷传递、材料强度和反应机理的关键因素。深入理解界面结构的性质对指导纳米复合材料的制备和应用具有重要意义。目前对于Al/Fe2O3纳米铝热剂界面的研究,多是通过电镜扫描观察Al 纳米颗粒与Fe2O3基体间的晶面堆垛,而对Al/Fe2O3界面的性质,如堆垛关系、界面强度和电子结构等仍不清楚。而第一性原理方法作为在电子水平提供基本信息的有力工具,可以对界面的电子结构进行定量预测,已成功应用于一些纳米铝热剂的计算研究,例如Shimojo 等[11-13]采用从头算分子动力学(AIMD)方法研究了Al/Fe2O3的电子结构性质,发现1ps 内在界面处发生氧化还原反应。Lanthony 等[14]采用密度泛函方法(DFT)研究了CuO沉积在Al(111)表面以及Al 沉积在CuO(11-1)表面的初始过程,探讨了Al/CuO 界面的生长机制。唐翠明 等[15]采 用AIMD 研 究 了2000 K 下Al/Fe2O3的 铝 热反应,结果表明反应过程中化学键和电荷量随时间的增加而变化。Xiong 等[16]采用DFT 方法研究了不同金属与CuO(111)构成的界面,分析了不同铝热界面的电 子 性 质 和 稳 定 性。Feng 等[17]采 用AIMD 模 拟 了Al/NiO 纳米铝热剂点火和燃烧反应过程,发现一次放热和二次放热分别是由界面反应和块体反应引起。但是,目前尚未有关于Al/Fe2O3界面性质的理论研究。
本研究采用周期性DFT 方法研究了Al(111)/Fe2O3(104)和Al(111)/Fe2O3(110)铝热剂界面的黏附特性和电子结构,其中Al(111)和Fe2O3的(104)和(110)表面是X 射线衍射实验中的主要暴露晶面[9],也是目前研究较多的晶面[18-19]。通过计算分析Fe2O3(104)和Fe2O3(110)的表面结构和表面能的大小,搭建了5 种Al/Fe2O3界面模型,研究了Al/Fe2O3铝热剂体系的界面结构、黏附功和电子结构。该工作可为Al/Fe2O3纳米铝热剂的材料设计、性能优化以及工程应用提供理论支持与指导。
2 计算方法
计 算 采 用 周 期 性DFT 方 法[20-21]和VASP 程 序包[22-24]完 成。 采 用 广 义 梯 度 近 似(GGA)下 的Perdew-Burke-Ernzerhof(PBE)[25]方 法 处 理 电 子 与 电子之间的交换关联势,采用投影缀加波方法(PAW)[26]描述离子与电子之间的相互作用,价电子在平面波中展开,平面波截断能设为450 eV,并对能量做收敛测试以保证结果准确可靠。由于α-Fe2O3是一个强关联反铁磁体系,包含局域的d轨道电子,因此采用了DFT+U 的修正方法,U 值选取根据文献报道对Fe 取4 eV[27]。Brillouin 区 数 值 积 分 采 用Monkhorst-Pack取样方案[28],对α-Fe2O3块体倒易空间网格点取5×5×5,Fe2O3(104)表面和Al(111)/Fe2O3(104)界面取3×3×1,Fe2O3(110)表面和Al(111)/Fe2O3(110)界面取3×5×1。为避免界面层之间周期性作用的影响,表面设置厚12 Å 的真空层。电子自洽迭代能和弛豫过程原子上的力的收敛标准分别为10-4eV 和0.03 eV·Å-1。所有计算均考虑自旋极化效应。基于以上参数设置,对α-Fe2O3和Al 晶体进行了测试,优化得到的晶胞常数(Fe2O3为a=b=5.061 Å,c=13.835 Å;Al 为a=b=c=4.041 Å)与实验值(Fe2O3,a=b=5.038 Å,c=13.772 Å;Al,a=b=c=4.05 Å)[29-30]以及文献报道的理论计算结果(Fe2O3,a=b=5.066 Å,c=13.868 Å;Al,a=b=c=4.04 Å)[31,18]相 近,表 明 所 用 计 算 方 法 和 参 数 是 可靠的。
3 结果与讨论
3.1 表面结构
在研究界面结构前先讨论构成界面的表面结构。Fe2O3(104)表面有两种不同类型的结构:一种是表层只有O 原子暴露(记为FS1,见图1),另一种是表层有Fe 和O 两种原子暴露,且亚表层也是Fe 和O(FS2),或亚表层只有O(FS3)。Fe2O3(110)表面也有两种不同类型的结构:表层只有O 原子暴露且亚表层为Fe(FS4)或亚表层为O(FS5),以及表层只有Fe 原子暴露(FS6)。因此,共构建了7 种上下对称的表面结构,即Al(111)和FS1~FS6,其中FS3 和FS4 表面符合化学计量比,FS1、FS2、FS5 和FS6 表面不符合化学计量比。
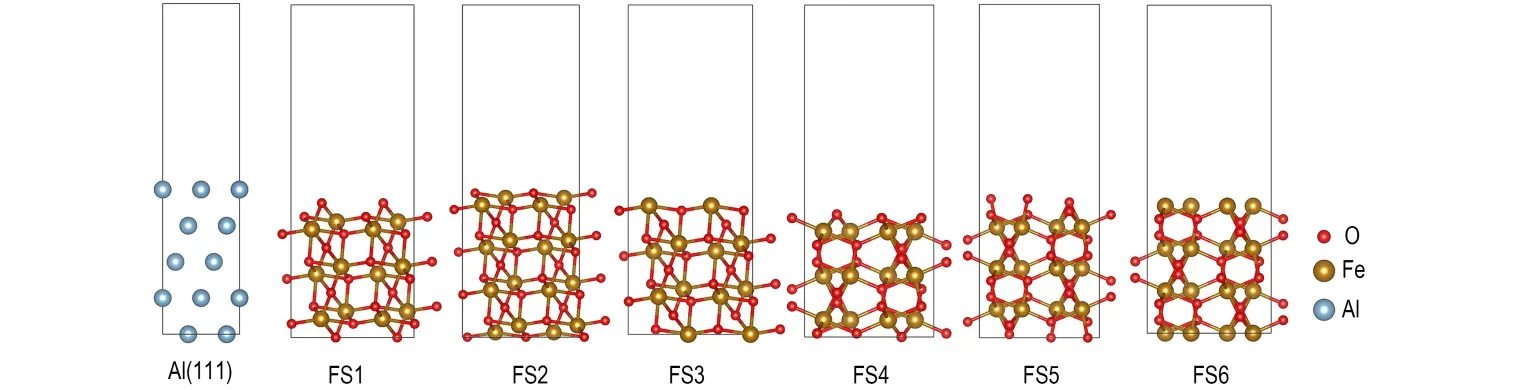
图1 Al 和Fe2O3的表面结构Fig.1 Surface structures of Al and Fe2O3
为保证计算结果可靠,须测试每个表面模型的厚度。通过计算不同原子层数的表面模型的表面能(γ),可以对表面γ模型进行收敛性测试,也就是确定合适的表面模型的厚度,使表面模型足以表现块体性质。γ由公式(1)计算[32-33]:

式中,Gslab是表面模型的吉布斯自由能[30],J·mol-1;Ni是与表面模型同样大小的块体中i原子的数量,无量纲;μi是i原子的化学势,J·mol-1;由于模型有两个对称表面,故表面积为2A,m2·mol-1。在平衡条件下,块体的化学势μbulk等于块体的摩尔吉布斯自由能,即单位块体中每种组分的化学势之和,因此对于Al(111)和Fe2O3表面分别有式(2)和(3):

根据吉布斯自由能的定义[30]:G=E+pV-TS(式中E是内能,J·mol-1;p是大气压,Pa;V是体积,m3;T是温度,K;S是熵,J·mol-1·K-1),由于pV和TS项对固体材料的G贡献很小,因此G近似等于能量E。故对于Al(111)和Fe2O3表 面,(1)式 可 分 别 化 为(4)和(5)式:

对于Al(111)表面,由公式(4)计算得到的结果表明,当原子层厚度n≥5 时,γ趋于固定值0.83 J·m-2,与文献报道的结果一致[34]。因此,本研究采用层数为5的Al(111)表面模型。对于Fe2O3(104)和Fe2O3(110)表面,由公式(5)计算得到不同n下FS1~FS6 的γ值列于表1。显然,对于表面FS1~FS6,当原子层数分别达到7、8、6、6、7、8 时,γ已基本趋于某个定值,表明在此厚度下这些表面模型已能较好表现体相性质,综合考虑计算精度和成本,对FS1~FS6 表面的厚度分别选取了n=10、11、9、9、10、11。另外,从表1 还可以发现,对这6 种不同的Fe2O3表面,其γ的大小次序为:FS4<FS1<FS2<FS3<FS6<FS5。在Fe2O3(110)的3 种暴露面中FS4 的γ最小,稳定性最好,与之前文献报道的结果一致[35],而FS5 和FS6 的γ比其它表面大得多,这主要是因为FS5 和FS6 表面的悬空键较多,表面稳定性较差。Fe2O3(104)的3 种暴露面的γ与FS1 差距不大,稳定性较好,这也与之前的报道相近[36]。因此,本研究后续计算只考虑较稳定的FS1~FS4 表面,其中前3 种为(104)表面,FS4 为(110)表面。
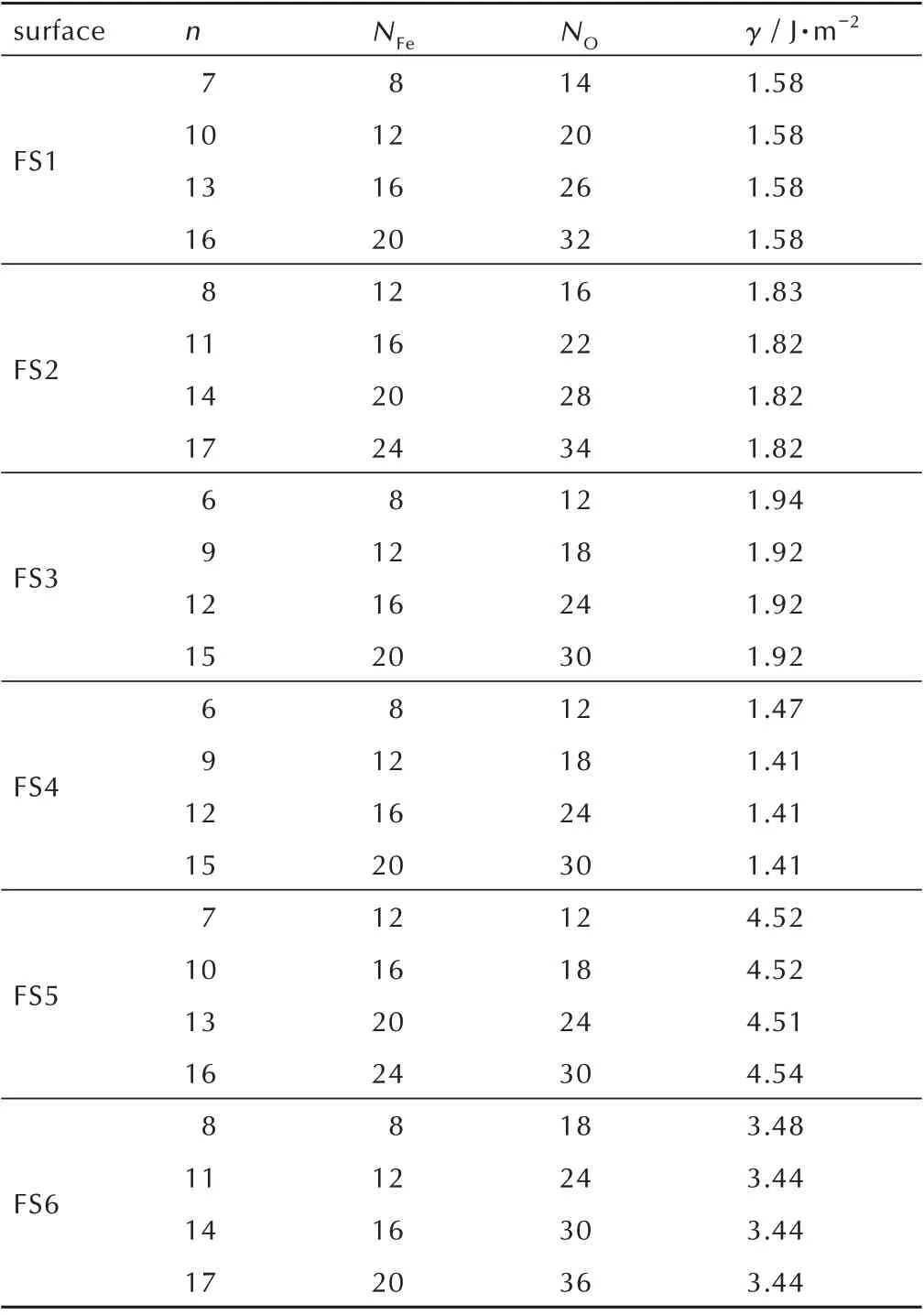
表1 不同层数的Fe2O3(104)和Fe2O3(110)表面模型的表面能Table 1 Surface energies of Fe2O3(104)and Fe2O3(110)surfaces with different number of layers
3.2 Al/Fe2O3界面
3.2.1 界面几何结构
在对Al 和Fe2O3表面模型的厚度及稳定性的测试结果基础上,构建了Al(111)/Fe2O3界面结构模型。构建界面模型时,对两个表面均引入了一定的晶格畸变。对于Al(111)/Fe2O3(104)界面,在Al 块体上沿[111]方向截取(3×3)的二维Al(111)平板(晶格常数μ=ν=8.59 Å)堆积在Fe2O3(104)(晶格常数μ=ν=9.0 Å)上构建界面模型,构建的界面模型的晶格常数(μ=ν=8.80 Å)与Al(111)和Fe2O3(104)晶格常数的相对误差Δμ和Δν均小于2.34%。同样,对于Al(111)/Fe2O3(110)界面,截取(3×2)的Al(111)平板(晶格常数μ=8.59 Å,ν=5.73 Å)堆积在Fe2O3(110)(晶格 常数μ=8.77 Å,ν=5.46 Å)上构建界面模型,构建的界面模型的晶格常数(μ=8.68 Å,ν=5.59 Å)与Al(111)和Fe2O3(110)晶格常数的相对误差Δμ和Δν也都小于2.39%。由于Fe2O3(104)和Fe2O3(110)表面有不同暴露结构且Al(111)表面的Al 原子在Fe2O3表面还有不同的堆积位置,由FS1~FS4 与Al(111)表面依次构建了5 种可能的界面结构模型,分别为AFS1、AFS2、AFS3、AFS4 和AFS5(见图2)。其中,AFS4 和AFS5 均由FS4 表面与Al(111)表面构成,分别对应于Al 原子堆积在FS4 表面的两种不同位置,即O 原子空位和O 原子顶位。在界面结构模型的弛豫过程中,由于距离接触界面远的原子位置变化很小,因此,对距接触界面最远的Fe2O3表面的4 层和Al(111)表面的2 层进行了固定。
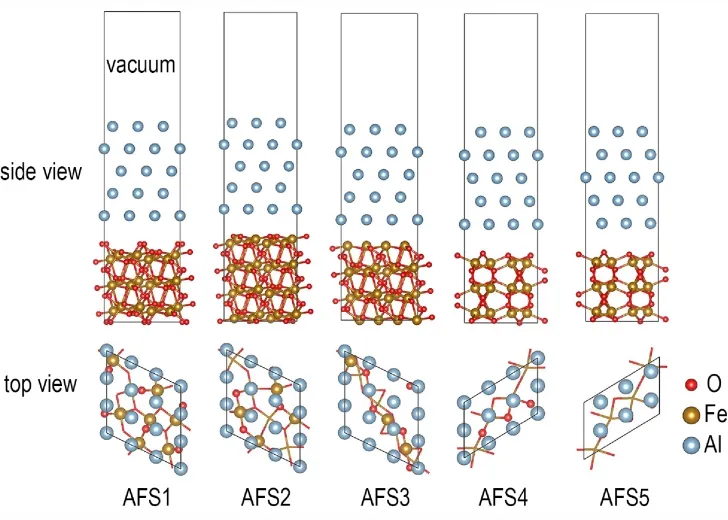
图2 5 种Al/Fe2O3界面结构Fig.2 Five interface structures of Al/Fe2O3
图3 是弛豫后的界面结构。弛豫前后界面处Al原子位置发生了较大变化,在垂直于和平行于界面方向都有移动,界面间距减小。在AFS2 和AFS3 界面中,Al 原子的紧密堆叠导致Fe2O3表面重构,Fe2O3表面O 原子垂直于界面向Al 表面移动,形成了与AFS1 中相似的O 原子暴露界面结构,说明AFS1 比AFS2 和AFS3 界面更稳定。在AFS4 界面中,界面处Al 原子在弛豫过程也发生了移动,从O 原子空位移动到O 原子顶位,形成了与AFS5 相似的界面结构,说明O 原子顶位堆叠的AFS5 界面比AFS4 界面更稳定。
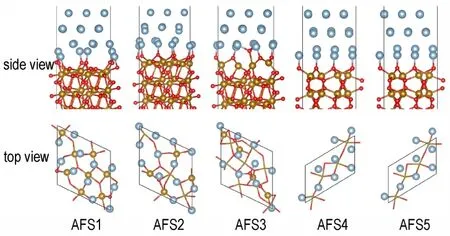
图3 5 种Al/Fe2O3界面的弛豫结构Fig.3 Five relaxed interface structures of Al/Fe2O3
3.2.2 界面黏附功
黏附功(Wad)是将一个界面分离为两个自由表面所需的单位面积可逆功,因此可用来评价界面的结合强度和稳定性。Wad越大,界面原子间的结合力越强,界面越稳定。Wad由式(7)求得[37]:

式中,EAl和EFe2O3分别是界面中Al 和Fe2O3表面的总能量,Eintf是Al/Fe2O3界 面 的 能 量,J·mol-1;A是 界 面 面积,m2·mol-1。
表2 列 出 了Al(111)/Fe2O3(104)和Al(111)/Fe2O3(110)界 面AFS1~AFS5 的Wad。从表中可以看出,Al(111)/Fe2O3(104)界 面 的 黏 附 功 明 显 大 于Al(111)/Fe2O3(110),说明Al(111)/Fe2O3(104)界面比Al(111)/Fe2O3(110)界面更稳定。与其他体系相比[16],金属层与Fe2O3的相互作用更强,黏附功更大,也预示着界面存在较强的离子特性。对于Al(111)/Fe2O3(104)界面,三种界面结构模型的黏附功分别为3.92、3.27 和3.43 J·m-2。AFS1 界面的黏附功最高,说明O 原子暴露的Fe2O3(104)表面与Al 原子的结合强度更高,界面结构更稳定。AFS2 和AFS3 界面的黏附功小于AFS1,结合强度低于AFS1,界面结构稳定性相对较差。对于Al(111)/Fe2O3(110)界面,O 原子顶位堆积的AFS5 界面黏附功为3.02 J·m-2,略高于空位堆积的AFS4 界面的黏附功(2.98 J·m-2),说明顶位堆叠的AFS5 界面结合强度更高更稳定。Wad的结果与弛豫过程结构的变化规律吻合。
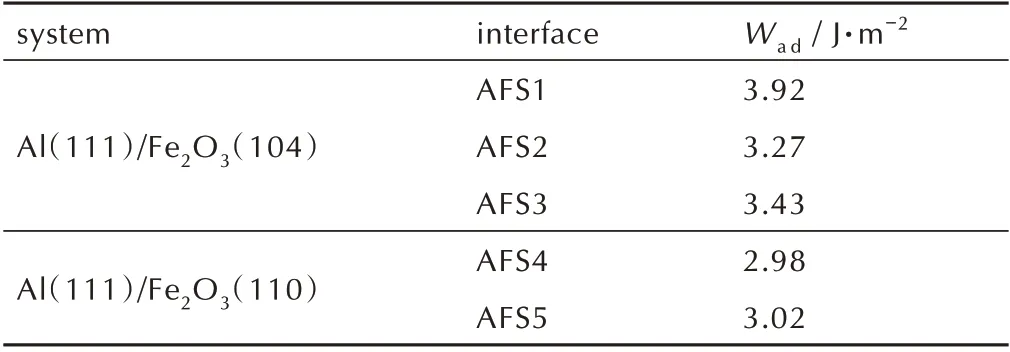
表2 5 种界面的黏附功(Wa d)Table 2 Adhesion work(Wad)of five Al/Fe2O3 interfaces
3.2.3 界面电子结构
为了揭示界面的电子结构和键合特征与界面稳定性的关系,由公式(8)计算了Al(111)/Fe2O3(104)和Al(111)/Fe2O3(110)界面中Wad最大的AFS1 和AFS5的差分电荷密度,还采用平面平均法计算了垂直于界面(即z轴)方向的平均差分电荷密度(见图4,图中黄色和蓝色分别表示电荷密度的增加和减少)。

式 中,ρintf、ρAl和ρFe2O3分 别 是Al/Fe2O3界 面、Al 表 面 和Fe2O3表面的电荷密度,e·Å-3。由图4a 和图4b 可见,在AFS1 界面中,Fe2O3(104)表面O 原子与Al(111)表面Al 原子之间有大量电荷聚集,Al 原子失去部分电荷而O 原子得到部分电荷,Al 和O 原子之间有较强的静电作用,在界面处形成Al-O 离子键,因此AFS1 界面主要是通过Al─O 离子键作用形成。根据图4c 和图4d,对于AFS5 界面,界面处也存在很多自由电子,主要来源于Fe2O3(110)表面的O 原子和Al(111)表面的Al 原子,说明该界面处同样存在较强的静电作用,形成了Al─O 离子键,因此AFS5 界面也主要是通过Al─O 离子键作用形成。比较AFS1 和AFS5 界面的价电荷相互作用和电荷聚集程度,前者明显强于后者,这与前者的黏附功更大的结果相吻合。

图4 AFS1 界面(a,b)和AFS5 界面(c,d)的差分电荷密度和平面平均差分电荷密度(黄色和蓝色分别表示电荷密度的增加和减少)Fig.4 Differential charge density and plane-averaged difference charge density of AFS1(a,b)and AFS5(c,d)(Yellow and blue colors represent the increase and decrease in charge density,respectively)
为更深入了解界面的电子结构,还计算了AFS1 和AFS5 界面的分波态密度(PDOS)(见图5),图5 给出了界面处O 和Al 以及远离界面的内层O 和Al 原子的PDOS,内层O 和Al 原子视作为体相原子,可以看出,界面处原子的PDOS 与内层原子不同,说明界面处发生了电 荷重新分布。在AFS1 界面上,O 原子的p轨道在-6 eV 处出现了新的峰,且峰值与Al 原子的峰值相近,主要是O-2p和Al-3s之间的杂化,说明Al(111)/Fe2O3(104)界面中形成了Al─O 离子键。与体相原子相比,界面处Al 原子在费米能级处的占据态较低,进一步说明了界面上形成了Al─O 离子键。在AFS5界面上,O 原子也在-6 eV出现新的峰,且峰值也与Al(111)表层Al 原子相近,说明Al(111)/Fe2O3(110)界面处也有Al─O 离子键形成。同时,与体相Al 原子相比,表层Al 原子的PDOS 曲线更平缓,特别是费米能级处的占据态更低,进一步说明界面处有Al─O 离子键形成。
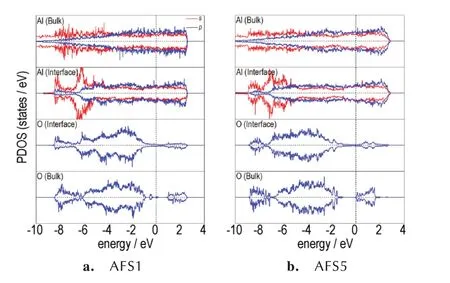
图5 Al/Fe2O3 界面的分波态密度图:(a)AFS1 界面;(b)AFS5界面Al(Bulk)、O(Bulk)、Al(Interface)、O(Interface)分别表示内层(体相)及界面Al 和O 原子Fig.5 The partial density of states of Al/Fe2O3 interfaces:(a)AFS1 interface;(b)AFS5 interface. Al(Bulk),O(Bulk),Al(Interface),and O(Interface)represent the inner Al and O atoms and interface Al and O atoms,respectively.
4 结论
采用周期性密度泛函理论方法研究了Fe2O3(104)和Fe2O3(110)表面以及Al/Fe2O3界面的性质,分析了不同表面和界面的结构、表面能和界面黏附功。结果表明:
(1)O原子暴露的Fe2O3(104)表面比其它Fe2O3(104)表面更稳定,同样地,O 原子暴露且亚表层为Fe 原子的Fe2O3(110)表面比其它Fe2O3(110)表面更稳定。
(2)在5 种Al/Fe2O3界面中,由O 原子暴露表面形成 的Al(111)/Fe2O3(104)界 面AFS1 和Al(111)/Fe2O3(110)界面AFS5 具有较大的界面黏附功,黏附功分别为3.92 J·m-2和3.02 J·m-2,界面较稳定。
(3)在这两种界面结构中,电荷转移主要发生在界面处的Al 和O 原子层之间。界面处有Al─O 离子键形成,且轨道电子的杂交来源于Al-3s轨道和O-2p轨道。
猜你喜欢
物理通报(2024年4期)2024-04-09 12:41:28
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 06:42:32
中学生数理化·中考版(2021年10期)2021-11-22 07:26:40
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27 03:07:02
高中数理化(2020年16期)2020-10-14 11:51:52
考试周刊(2019年2期)2019-01-28 10:08:56
新高考·高一物理(2015年6期)2015-09-28 20:10:57
化学教与学(2014年12期)2014-12-12 10:05:09
