
羊口疮病毒F1L 蛋白对山羊痘病毒黏附BHK21 细胞的影响
2022-03-08 06:46杨柳许国洋牟豪白运川余远迪
湖南农业大学学报(自然科学版) 2022年1期
杨柳,许国洋#,牟豪,白运川,余远迪*
(1.重庆市畜牧科学院,重庆 402460;2.重庆市兽用生物制品工程技术研究中心,重庆 402460;3.重庆酉阳土家族苗族自治县畜牧产业发展中心,重庆 409812)
山羊痘、羊口疮广泛发生于养羊的国家(地区),对牧羊造成严重危害,给农牧民带来较大经济损失。近些年来,随着养羊业的发展和生产方式的改变,羊病发生日趋复杂。临床上既有单一感染山羊痘病毒(GTPV)或羊口疮病毒(ORFV)的病例[1-2],也存在2种病毒混合感染发病的情况[3]。由于羊口疮和羊痘临症相似,导致诊断困难,常常出现误诊,鉴别诊断羊口疮或羊痘一直是当前研究热点[4]。GTPV AV41株弱毒活疫苗在中国已应用多年,在防控山羊痘的过程中发挥了关键性作用,其安全性和有效性已得到充分证明。然而羊口疮在羊场常见多发,目前中国无商品化的羊口疮疫苗。当羊群携带ORFV时注射山羊痘活疫苗,其免疫效果是否受影响还有待深入研究。
F1L蛋白是ORFV的一种重要囊膜蛋白,为ORFV的059基因编码。CZERNY等[5]用ORFV的单克隆抗体通过竞争性ELISA、免疫金电镜技术和Western blot等方法对病毒囊膜上的多种蛋白进行研究,发现单克隆抗体能与ORFV病毒表面特异性的抗原决定簇结合,其中相对分子质量大约3.9×104的病毒表面蛋白就是单克隆抗体的主要结合位点。根据DELHON等[6]、MERCER等[7]发布的ORFV全基因组DNA序列,分析发现3.9×104蛋白为ORFV基因组的中心保守区ORFV 059基因编码产生的,该基因编码区DNA长约1023 bp,包含1个完整的开放阅读框(ORF)。SCAGLIARINI等[8]进一步鉴定ORFV F1L蛋白是免疫优势蛋白,免疫金染色显示该蛋白位于ORFV的表面。GALLINA等[9]克隆、表达了F1L基因,纯化蛋白免疫家兔,结果显示F1L蛋白有较强的免疫原性,能刺激宿主产生针对ORFV的中和抗体,意味着F1L蛋白具有作为亚单位疫苗的潜力。尤其是SCAGLIARINI等[10]发现了肝素对F1L蛋白与胎羊睾丸细胞结合有竞争性抑制作用,证明ORFV F1L蛋白具有肝素结合活性,能够与表达于大多数哺乳动物细胞表面的硫酸乙酰肝素受体结合,故认为该蛋白在病毒吸附和侵染细胞的早期阶段发挥了重要作用。由于F1L蛋白既能够诱导机体产生保护性中和抗体,还可以阻止病毒ORFV吸附宿主细胞而发挥免疫作用,从而利于机体清除病原体,认为F1L蛋白已成为研制ORFV新型亚单位疫苗靶标抗原[9]。已有的资料[11]显示,黏附宿主细胞是病原感染发生的关键。许多病原微生物产生黏附蛋白,以促进其黏附宿主细胞。以GTPV AV41株研制的活疫苗免疫接种山羊后,病毒黏附侵染宿主细胞,在细胞内有一定程度的繁殖或复制,激发机体对病原的持久免疫力。目前,还少见F1L蛋白影响GTPV黏附细胞的相关报道。
本研究中,以ORFV F1L为配体蛋白、GTPV为靶标病毒、乳仓鼠肾成纤维细胞(BHK21)为宿主细胞,将蛋白F1L、病毒GTPV作不同处理后孵育BHK21细胞,测定孵育细胞黏附GTPV的量,分析GTPV黏附细胞拷贝数的变化,探究ORFV F1L蛋白是否干扰GTPV黏附BHK21细胞和ORFV对GTPV感染山羊细胞的影响,旨在为生产实际中用GTPV弱毒活疫苗免疫羊群提供依据。
1 材料与方法
1.1 材料
1.1.1 供试细胞和病毒
BHK21 细胞为重庆市兽用生物制品工程技术研究中心保存;GTPV 疫苗弱毒株(GTPV AV41 株)为重庆澳龙生物制品有限公司惠赠,测定病毒TCID50约为1×10-4/mL。
1.1.2 主要试剂和引物
病毒RNA/DNA 提取试剂盒(Viral RNA/DNA Extraction Kit Ver5.0)、DNA 胶回收试剂盒、质粒提取试剂盒购自OMEGA;琼脂糖粉、5-溴-4-氯-3-吲哚-β-D-吡喃半乳糖苷(X-gal)、异丙基硫代半乳糖苷(IPTG)、限制性内切酶Hind III、EcoR I、T4DNA ligase、PCR Master Mix(2×)、pMD18-T vector 购自TaKaRa;DMEM 细胞培养液购自Gibco;驴抗羊荧光标记二抗购自Invitrogen。参考GenBank 公布的基因(ID 为KX951408.1),设计合成ORFVF1L基因 引 物(5'-ATGGATCCACCCGAAATCACG-3',5'-TCACACGATGGCCGTGAC-3');参考GenBank公布的基因(ID 为AY040083.1),设计合成GTPVP32基因引物(5'-ATGGCAGATATCCCATTA-3',5'-CTAAACTATATACGTAAATAAC-3');参考文献[12],设计合成荧光定量引物(5'-TGTTATTATGT TTGATCCCGTTC-3',5'-GTAAAATCATATAAGG TGCGACAA-3')。
1.1.3 主要仪器
气浴振荡器为上海博迅实业有限公司的产品;细菌培养箱为上海跃进医疗器械有限公司的产品;台式高速冷冻离心机为Heraeus 的产品;凝胶成像系统为Syngene BOX EF 的产品;电泳仪为上海比朗仪器制造有限公司的产品;细胞培养箱为Thermo的产品;倒置显微镜为Olympus 的产品;低速离心机HICO21 为生工生物工程(上海)股份有限公司的产品;PCR 仪为ABI VeritiTM的产品;Odssay®CLX为Gene Company Limited 的产品;核酸蛋白浓度测定仪为Eppendorf BioPhotometer 的产品。
1.2 方法
1.2.1 病料处理和病毒DNA 的提取
采集重庆市石柱某羊场发病羊的痂皮并称量痂皮的质量,加入5 倍量的磷酸缓冲溶液(1×PBS,pH 7.2),用研钵反复研磨;收集研磨液,反复冻融3 次,3000 r/min 离心30 min;上清用0.45 μm 滤器过滤,收集滤液,-20 ℃保存,备用。同时取GTPV AV41 株病毒溶液、羊口疮病样滤液各200 µL,采用试剂盒提取病毒DNA,并将其置于-20 ℃冰箱保存,备用。
1.2.2 ORFV 059 基因的扩增和克隆及测序
以提取羊口疮病料的DNA 为模板,PCR 扩增ORFV 059 基因。反应体系(50 µL):2×TaqPCR Master Mixes 25.0 µL,DNA 4.0 µL,上游引物(10µmol/L) 1.0 µL,下游引物(10 µmol/L) 1.0 µL,ddH2O 19.0 µL。反应条件:94 ℃预变性5 min;94℃变性1.0 min,55 ℃退火45 s,72 ℃延伸1.5 min,共30 个循环;72 ℃再延伸10 min。PCR 产物用1×TAE Buffer、1.0%琼脂糖凝胶在120 V 条件下电泳,EB 染色观察,切胶回收目标DNA,连接pMD18-T vector,构建重组质粒,转化感受态Escherichia coliDH5α 后涂布培养。采用蓝白斑法挑取阳性菌落培养,用试剂盒提取阳性菌的质粒并测序。
1.2.3 生物信息学分析
将测序获得的 DNA 序列在 NCBI 上进行BLAST 检索分析,运用在线软件(http://web.expasy.org/translate/)翻译F1L 蛋白,查找蛋白质序列中的碱性氨基酸,并分析蛋白保守结构域(http://smart.embl.de/smart/set_mode.cgi)、跨膜结构域和信号肽(https://phobius.sbc.su.se/)序列。
1.2.4 编码F1L 蛋白DNA 序列的优化及表达质粒的构建
根据大肠埃希菌密码子偏好性,对编码F1L 蛋白的DNA 序列作优化处理,在序列上、下游末端分别加EcoR I 和Hind III 单一酶切位点,优化序列送至生工生物工程(上海)股份有限公司合成。采用EcoR I、Hind III 双酶切质粒pET42a(+),优化合成DNA 片段,酶切产物用1.0%琼脂糖凝胶电泳,切胶回收目标片段。将回收片段(带黏性末端优化DNA 和载体pET42a(+))用T4DNA ligase 于16 ℃环境中连接过夜,连接产物转化感受态E. coliBL21(DE3)后涂板培养。挑单克隆于LB 试管中振摇过夜,取培养液进行菌液PCR,产物经电泳检测,对阳性克隆菌的质粒测序鉴定。
1.2.5 蛋白的表达及免疫印迹试验
挑取阳性单克隆菌并接入LB培养液中,于37℃下180 r/min振摇培养。当菌液OD600nm=0.4时,加入终浓度为0.1 mmol/L的IPTG,同时培养空质粒pET42a(+)菌作为对照。分别于3、6 h取诱导菌液大约1.0 mL,于4 ℃下5000 r/min离心10 min后去上清;用1×PBS混匀沉淀,5000 r/min离心10 min之后弃上清,重复洗涤3次;最后向沉淀中加100 µL蛋白上样缓冲液并吹打混匀,于沸水中煮沸10 min后冷却,用SDS-PAGE胶(5%浓缩胶+12%分离胶)电泳。一方面,将电泳胶经考马斯亮蓝染色之后脱色观察,初步检测蛋白是否表达;另一方面,对电泳胶进行转移电泳,将胶中蛋白转移到膜上,以ORFV抗体(羊口疮阳性血清)为一抗(1∶500)对转移膜进行孵育,再用1×PBS洗涤后用驴抗羊荧光标记二抗(1∶5000)进行孵育,最后用1×PBS洗涤后采用ODssay成像仪对孵育膜摄像,进一步检测表达蛋白是否为F1L蛋白。
1.2.6 蛋白的纯化和复性及浓度测定
收集IPTG诱导的重组质粒菌体,用20 mL 1×PBS重悬1000 mL LB过夜培养的菌体,再在冰水中超声裂解菌悬液(功率300 W,工作4 s、间歇5 s,超声裂解30 min);裂解液以12 000 r/min离心10 min,收集上清和沉淀,分别作SDS-PAGE电泳检测。对于包涵体表达的蛋白,裂解沉淀用pH 8.0的缓冲液(8 mol/L尿素、20 mmol/L Tris-HCl、2 mmol/L EDTA、2 mmol/L巯基乙醇)溶解过夜后,12 000 r/min离心10 min,收集上清进行SDS-PAGE电泳观察。
参照文献[13]的方法,采用His柱纯化蛋白,再用SDS-PAGE分析蛋白纯化效果。参照文献[14]的梯度透析复性方法,对过柱纯化的蛋白质作复性处理。按照Bradford蛋白浓度测定试剂盒说明书测定复性蛋白质的浓度。
1.2.7 GTPVP32基因克隆及其绝对定量标准曲线制定
以提取GTPV AV41 株基因组DNA 为模板,通过PCR 扩增P32基因,产物用1.0%的琼脂糖凝胶电泳,切胶回收目标条带,产物连接pMD18-T 载体、转化E. coli;挑阳性克隆菌作液体培养,再对菌液进行测序鉴定;采用试剂盒抽提pMD18-P32质粒,以此为模板作梯度稀释,参照文献[12]的方法,制定GTPV 绝对定量标准曲线。
1.2.8 F1L 蛋白对GTPV 黏附BHK21 细胞试验
从液氮中取出BHK21 细胞冻存管,快速置于37 ℃的水中,不停摇动直至全部融化;将融化液加到有10%小牛血清DMEM 液的细胞瓶,于37 ℃5% CO2培养箱中培养;待细胞长满瓶后弃细胞培养液,用1×PBS 洗涤,再以胰酶消化适当时间;取完全培养液终止消化,将贴壁细胞轻轻吹打下来,低速离心收集细胞;用10%小牛血清DMEM 培养基重悬细胞,将重悬液加至12 孔细胞板中,然后置于细胞培养箱中培养,直至细胞长至单层。用1×PBS 洗涤BHK21 单层细胞,再用0.15 mg 复性的蛋白F1L、0.15 mL 病毒GTPV 对单层细胞进行蛋白孵育(阴性对照)、先蛋白孵育后病毒孵育、蛋白和病毒混液孵育、病毒孵育(阳性对照)4 种处理。每种处理设置3 个重复。参照文献[15],选择病毒DNA 于细胞内未复制前,即病毒孵育1.0、6.0 h 终止孵育;用1×PBS 洗涤3 次,之后每孔加200 µL 1×PBS 反复冻融;将冻融液体以12 000 r/min 离心10 min,取上清,抽提DNA 为模板,进行荧光定量PCR。反应体系(20.0 µL):上游引物(10 µmol/L)0.4 µL, 下游引物(10 µmol/L) 0.4 µL, TB Green Premix ExTaq10.0 µL,模板2.0 µL,dd H2O 7.2 µL。反应条件:95 ℃预变性30 s;95 ℃变性5 s,60 ℃退火30 s,共40 个循环。每个样重复3 次。最后分析病毒拷贝数差异。
2 结果与分析
2.1 F1L 基因序列分析结果
从图1可知,只有泳道3在约1000 bp处呈现1条与预期分子大小相符的DNA条带。对目标条带克隆和测序,结果获得1023 bp大小的DNA序列。在线BLAST比对结果显示,该序列与ORFV新疆分离株F1L基因相似性达100%,表明病料3含有ORFV,将该病毒株命名为重庆石柱分离株(ORFV-CQsz)。
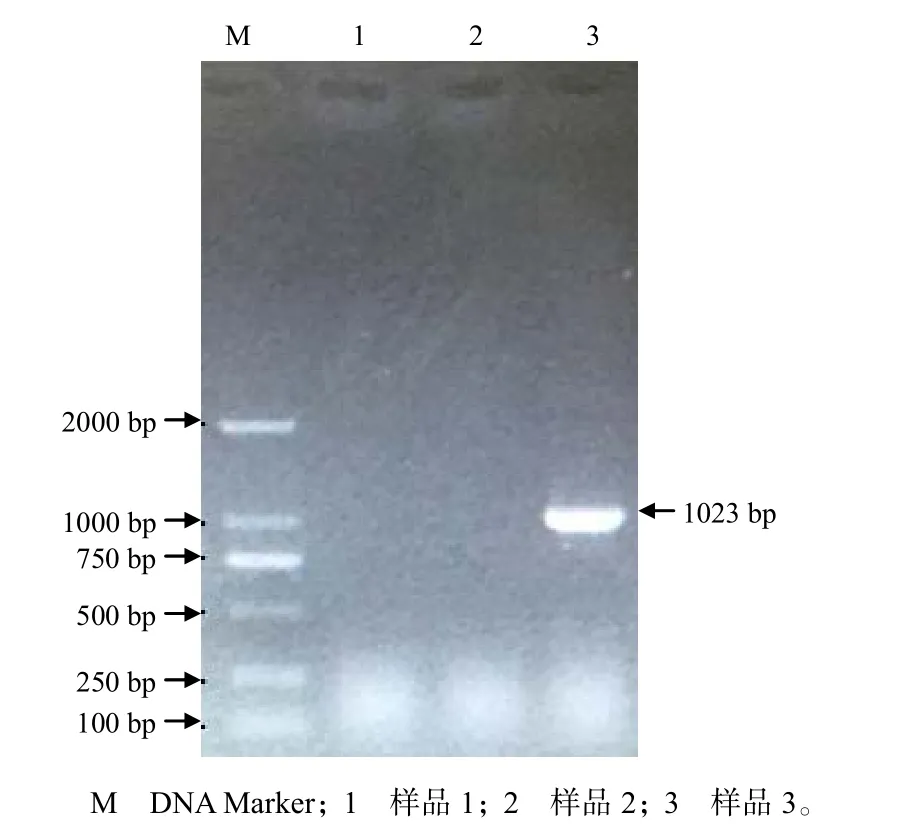
图1 ORFV F1L 基因的PCR 产物电泳结果Fig.1 Result of electrophoresis of PCR product of ORFV F1L gene
从图2可知,该DNA序列ORF编码340个氨基酸残基的蛋白质F1L,理论预测该蛋白的等电点为5.61,相对分子质量为3.73×104;在F1L蛋白中,共有11个R、19个K和5个H,碱性氨基酸含量为10.29%(35/340),并且其中有1段碱性氨基酸富集区;结构功能域分析显示,F1L蛋白包含1个类似Pox_P35结构域。该结构域的特点是能够与细胞表面带负电荷硫酸乙酰肝素(HS)受体结合,便于病毒黏附到细胞表面[16]。跨膜结构域和信号肽预测结果显示,蛋白F1L无信号肽序列,但构成蛋白的氨基酸序列C-端含有2段跨膜结构域。

图2 ORFV-CQsz F1L基因编码的氨基酸序列Fig.2 The amino acid sequence encoded by the ORFV-CQsz F1L gene
2.2 优化的编码F1L 蛋白DNA 序列及表达质粒鉴定结果
优化编码F1L 蛋白的DNA 序列与原编码的序列相比较,核苷酸变化的统计结果列于表1。与原序列相比,优化序列中的A、T、C、G 碱基占比趋向均匀,其中,GC 占比从原序列的64.8%调整到优化序列的50.0%。经PCR 检测及测序鉴定,基于优化序列构建的重组质粒阳性克隆菌均与预期的相符,结果表明成功获得优化DNA 序列构建的重组质粒菌。
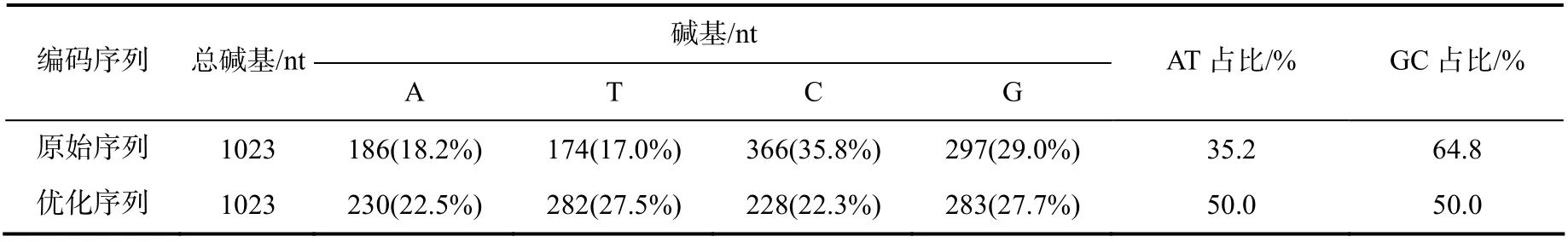
表1 编码F1L 蛋白DNA 序列优化前后的结果Table 1 Result of the DNA of F1L protein coding sequence before and after optimization
2.3 F1L 蛋白表达及免疫印迹结果
从图3可知,在相对分子质量大约为3.8×104处有1条明显的蛋白带,其相对分子质量大小与预期的相符;随着IPTG诱导时间延长,重组质粒菌表达的蛋白含量明显增加,初步证明该蛋白能被高效表达。从图4可知,在转移膜上可见相对分子质量约为3.8×104的条带,免疫印迹结果进一步证明表达的蛋白为F1L蛋白。
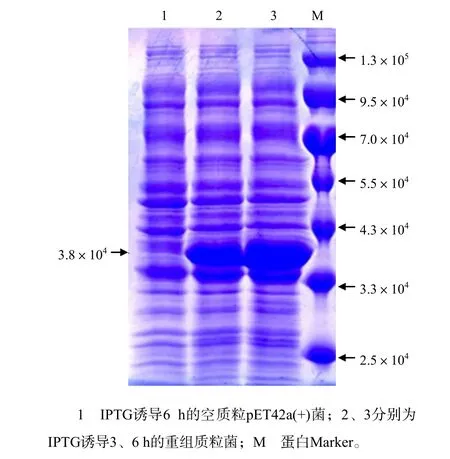
图3 F1L蛋白表达的SDS-PAGE结果Fig.3 Result of SDS-PAGE profile of expressed F1L protein
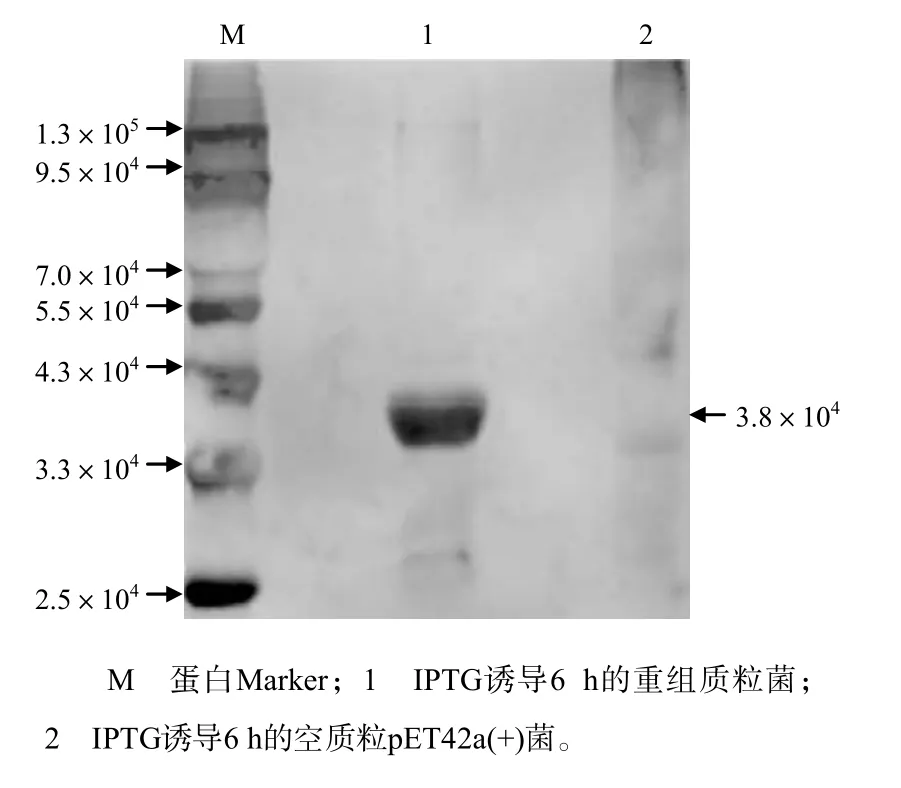
图4 F1L-CQsz蛋白免疫印迹检测结果Fig.4 Result of western blotting of F1L-CQsz protein
2.4 F1L 蛋白的纯化结果及复性蛋白质的浓度
对超声破碎IPTG 诱导重组菌悬液的SDSPAGE检测结果发现,裂解液上清液泳道中无明显可见的目的蛋白带,而沉淀泳道中出现相对分子质量约为3.8×104且与预期相符的蛋白带,表明F1L蛋白主要以包涵体的形式表达。对于用His柱纯化蛋白的检测,从图5可知,在相对分子质量大约为3.8×104处出现了1条明显的与预期相符的蛋白条带,表明纯化到了以包涵体形式表达的F1L蛋白。采用Bradford法测定的纯化复性的F1L蛋白质量浓度为1.06 mg/mL。
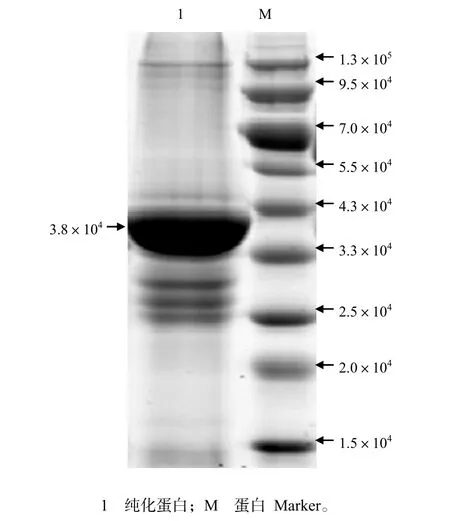
图5 纯化蛋白的SDS-PAGE分析结果Fig.5 Result of SDS-PAGE profile of the purified protein
2.5 GTPV P32 基因克隆及定量标准曲线
从图6 可知,在1000 bp 左右出现单一的DNA条带,该结果与预期分子大小相符。回收目的条带连接pMD18-T vector、转化E. coliDH5α,结果成功构建了重组质粒(pMD18-P32)菌。以提取的该质粒为模板,建立了研究GTPV AV41 株的绝对定量标准曲线(图7)。

图6 GTPV P32基因PCR产物电泳结果Fig.6 Result of electrophoresis of PCR product of GTPV P32 gene

图7 GTPV绝对定量标准曲线Fig.7 GTPV absolute quantitative standard curve
2.6 蛋白F1L 对GTPV 黏附BHK21 细胞的影响
从图8 可知,病毒GTPV 孵育1.0、6.0 h 的细胞,其黏附病毒数量均呈现为蛋白处理组的最低,病毒处理组的最高,先蛋白后病毒、蛋白和病毒混液共孵育的居中的特点。表明蛋白F1L 对GTPV 黏附到BHK21 细胞有一定的干扰作用,F1L 蛋白质降低了病毒黏附到细胞的数量。
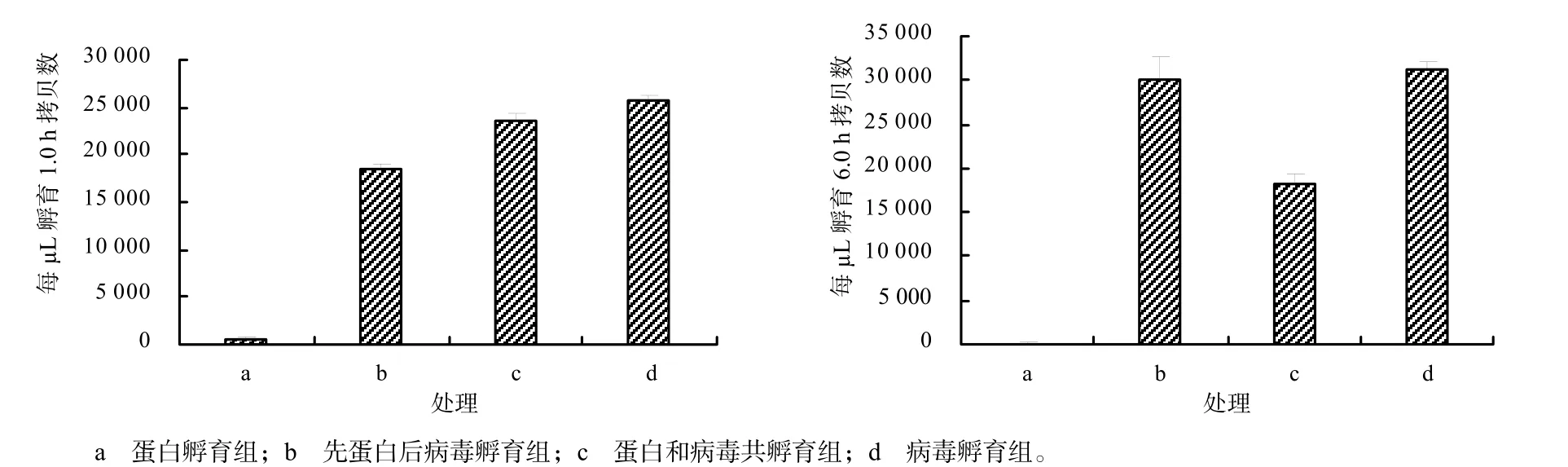
图8 荧光定量测定不同处理下细胞黏附GTPV 的拷贝数Fig.8 The copies of GTPV adhered to cells under different treatments were tested by fluorescence quantitative method
3 结论与讨论
本研究中,克隆了ORFV-CQsz 株的059 基因,发现该基因编码蛋白F1L的氨基酸序列C-端包含2个跨膜区域。由于存在跨膜区结构域,导致蛋白不容易获得高效表达。目前,对于跨膜蛋白的表达,通常采取截去跨膜区段,再对剩余部分蛋白质进行表达[17]。本研究中,通过对F1L-CQsz 蛋白质的DNA 编码序列进行优化处理后合成,以此构建原核表达质粒菌,最终实现了对目标蛋白的高效表达。
病原黏附细胞并侵入感染,通常是多种因子协同作用的结果[18],但黏附宿主细胞是感染发生的第一步。研究[19-21]发现,细菌、病毒、寄生虫等病原体特异地黏附真核细胞表面的氨基葡聚糖分子(如肝素、硫酸乙酰肝素、硫酸软骨素等多糖)受体,是形成病原感染的主要机制之一。经对ORFV-CQsz 059 基因及其编码蛋白分析,发现F1L-CQsz 蛋白有肝素结合活性;此外,作为一种痘病毒,GTPV外膜蛋白中包含有结合细胞表面肝素类似物的Pox_P35 结构域[16],这就意味着蛋白F1L-CQsz 和GTPV AV41 均可黏附细胞表面。在本研究中,对两者进行不同处理后孵育BHK21 细胞,结果发现F1L-CQsz 蛋白干扰了GTPV AV41 株黏附BHK21细胞,降低了病毒黏附细胞的拷贝数。其原因可能是蛋白与病毒竞争性吸附细胞表面受体,从而导致细胞黏附病毒的数量变少。本研究结果显示,F1L-CQsz 蛋白介导了ORFV 黏附BHK21 细胞,表明其功能可能与ORFV其他毒株的F1L蛋白功能一样,在黏附宿主细胞上发挥作用[10]。由于F1L 蛋白是ORFV 的一种重要囊膜蛋白,提示ORFV 可影响GTPV 黏附和感染宿主细胞。
鉴于国内尚无预防羊口疮的商品化疫苗,临床上对羊口疮的防控形势十分严峻。生产实践中以GTPV AV41 株制备的山羊痘活疫苗广泛免疫注射羊群,而ORFV 可影响GTPV 黏附和感染宿主细胞;因此,在使用GTPV 弱毒活疫苗免疫山羊之前,检测羊群健康状态,确保羊群不携带ORFV,可更好的发挥GTPV 活疫苗的免疫效果。
猜你喜欢
生殖医学杂志(2022年10期)2022-10-19
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
江西农业学报(2021年4期)2021-04-20
三农资讯半月报(2020年11期)2020-06-21
饮食保健(2020年10期)2020-05-29
中小学德育(2020年11期)2020-03-18
教育界·上旬(2016年12期)2017-05-25
莫愁(2017年12期)2017-04-22
标记免疫分析与临床(2016年9期)2016-11-21
