
进行性特发性皮肤萎缩1 例
2022-03-04 09:06刘宸序孔祥君聂振华詹菊芳
中国中西医结合皮肤性病学杂志 2022年1期
刘宸序,孔祥君,聂振华,詹菊芳
(1.天津中医药大学,天津 300193;2.天津市中医药研究院附属医院,天津 300120)
1 病例资料
患者女,18 岁,学生。因背部皮肤凹陷2 年余,于2021 年2 月1 日就诊于我院皮肤科。患者2 年前右侧背部出现暗红色斑片,无明显自觉症状。后斑片逐渐增大,颜色变暗,出现凹陷,皮损局部出现轻度萎缩,无炎性反应及硬化。既往体健,家族中无类似疾病史,个人史无特殊。体格检查:系统查体未见明显异常。皮肤科检查:右侧背部局限性萎缩性斑片,大小约8 cm×6 cm,与周围正常组织界限清晰,皮肤轻微凹陷,质地柔软,见图1。实验室检查:血、尿常规,免疫全项均未见明显异常。组织病理示:表皮基底层色素轻度增加,真皮内皮肤附属器上移,血管周围少数淋巴细胞浸润,见图2。弹力纤维染色:未见弹力纤维减少,见图3。
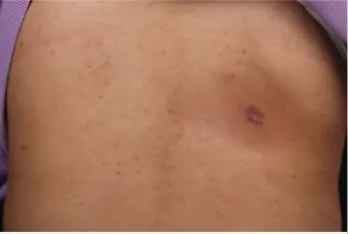
图1 患者皮损临床照片
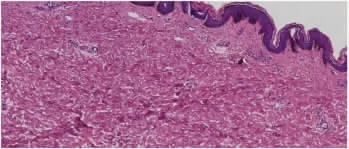
图2 皮损组织病理图(HE 染色×40)

图3 皮损弹力纤维染色图(弹力纤维染色×40)
诊断:进行性特发性皮肤萎缩(Atrophoderma of Pasini-Pierini,APP)。治疗:予复方丹参片2 片/次,3 次/d,口服,硬皮病颗粒、加减胃苓颗粒3 g/次,2次/d,口服。维生素E 乳膏、多磺酸粘多糖乳膏局部交替外用。经治疗1 个月,患者背部萎缩性斑片控制良好,未继续增大,目前随访中。
2 讨论
APP 又称“Pasini-Pierini 萎缩性皮病”是一种罕见的皮肤萎缩性疾病。其病因尚不明确,可能与伯氏疏螺旋体感染、神经源性病因、免疫因素和遗传易感性相关[1]。本病多发于青年女性,好发于躯干,尤以背部多见,也可累及四肢近端及其他部位。本病起病隐匿,常无主观症状,后期可引起疼痛、瘙痒,及感觉异常[2]。皮损单发或多发,常不对称,为圆形、椭圆形或不规则的萎缩斑,随病程进展逐渐扩大,色青紫或深棕,也可表现为色素减退[3]。皮损处表面光滑,轻微凹陷,与正常皮肤境界清楚,皮下静脉显露。缺乏炎性反应特征是本病的一个特征性表现[4]。病程呈慢性良性经过,可在数十年或更长时间中缓慢发展,最后呈静止状态,一般难以恢复。组织病理学早期变化轻微,表皮和真皮结缔组织变薄,真皮上部血管扩张并伴有稀疏的血管外炎细胞浸润[3],汗腺、毛囊皮脂腺和附属器表现正常。陈旧性皮损中可见表皮萎缩,真皮深层胶原束变粗、排列紧密呈纯化玻璃样变性,皮下脂肪层正常。因病理改变轻微,诊断需要与来自同一解剖部位的健康皮肤进行比较研究。除组织病理学检查外,高频和多普勒超声联合也可用于APP 诊断。在高频超声中,疾病早期可出现低回声和皮肤厚度增加,出现水肿表现,此特征可来区分硬斑病和APP;晚期皮肤厚度降低,回声增强,与纤维化和皮肤变薄相一致[5]。
本病是否为独立性疾病尚有争议,Jablonska等[6]认为本病为局限性硬斑病的一种特殊类型,局限性硬皮病在后期无论是临床特点还是组织病理特征均与APP 相似,皮损内有时可见局限性硬皮病样的硬化,同1 例患者可同时出现这2 种疾病,故有些学者认为APP 属于早期的硬斑病[3]。但APP 皮损先萎缩后硬化,与局限性硬皮病疾病发展顺序相反。此外,组织病理上局限性硬皮病可见单一核细胞在附属器周围浸润、附属器结构减少或消失,也有别于APP。弹性纤维在APP 中或减少或聚集,而局限性硬皮病的弹性纤维通常无明显变化。除硬斑病以外,APP 还应当与以下疾病相鉴别:①斑状萎缩,皮损处皮肤菲薄,呈蓝白色,可微隆起,触之有疝囊感,组织病理有中度血管周围炎及炎性细胞浸润,可见到弹力纤维不同程度的断裂和消失。②血管萎缩性皮肤异色症,多见于面部、四肢,呈对称分布,境界不清,可见红斑、脱屑斑、斑驳色素沉着、毛细血管扩张和皮肤萎缩,本病可作为多种炎性疾病,遗传病及肿瘤的表现。组织病理可见基底细胞液化变性、真皮上部炎性细胞呈带状浸润,胶原纤维正常。③皮下脂肪萎缩,仅皮下脂肪萎缩,表皮凹陷,但表皮厚度及色泽均正常。组织病理变化主要为皮下脂肪减少或缺乏。
APP 尚无特效的治疗方法,可口服丹参酮、维生素E 等药物进行治疗,针对伯氏疏螺旋体感染患者用四环素或青霉素治疗有一定作用[7],抗疟药例如羟氯喹,对同时患有系统性红班狼疮(SLE)的患者有良好的效应。此外,局部外用糖皮质激素、钙调磷酸酶抑制剂或维生素E 乳膏,局部调Q 翠绿宝石激光治疗也起到一定作用[8-9]。本例患者病理确诊后应用复方丹参片、中药颗粒口服,配合维生素E 乳膏、多磺酸粘多糖乳膏局部交替外用治疗,1 个月后随访皮损未继续扩大,患者目前仍在继续随访中。
猜你喜欢
中国典型病例大全(2022年9期)2022-04-19
现代仪器与医疗(2022年1期)2022-04-19
现代仪器与医疗(2021年5期)2021-12-02
婚育与健康(2021年14期)2021-10-18
科学之谜(2020年11期)2020-01-13
中外医学研究(2018年22期)2018-10-12
家庭医药(2018年2期)2018-02-09
中国医药导报(2017年10期)2017-06-01
浙江中医杂志(2004年1期)2004-07-30
浙江中医杂志(2004年5期)2004-03-09
