
锰系锂离子筛的制备与改性的研究进展
2022-02-21 03:36赵元元陈海峰刘云云吴勇民张竞择汤卫平
无机盐工业 2022年2期
赵元元 ,陈海峰 ,刘云云 ,张 宏 ,吴勇民 ,张竞择 ,汤卫平
(1.陕西科技大学机电工程学院,陕西西安 710021;2.上海空间电源研究所,空间电源技术国家重点实验室)
锂是现代能源发展的重要元素[1],这种最轻的碱金属广泛地应用于新能源等各个领域,是高比能量密度锂电池必不可少的原料,而锂电池又是新能源汽车、电网储能、便携式电子设备、电动工具的重要动力来源[2]。 近年来,随着新能源产业的快速发展,锂电池的需求呈爆发式增长,从而引发全球对锂资源的市场需求的不断增加[3]。
全球锂资源主要存在两种形式: 一是含锂矿石,如锂云母、锂辉石、锂皂石;二是含锂水资源,如盐湖卤水和海水[4-5]。 有调查表明,超过锂总量的60%的锂存在于盐湖和海水中[6]。 所以盐湖锂资源成为锂电池产业发展的重要来源。 盐湖提锂的方法有吸附法、电渗析、电化学脱嵌、溶媒萃取等[7-10]。 但除了南美的低镁锂比优质盐湖已经大规模开采外,大部分高镁锂比的盐湖锂资源的开采遇到困难。 由于 Mg2+和 Li+之间有着几乎相同的性质[11-12],对镁锂物质的量比超过6 的盐湖卤水, 常规的太阳能蒸发沉淀法的应用就非常有限[13],而吸附法就为解决这一难题提供了可能性。
吸附法中所用的吸附剂又可分为铝系吸附剂和锰系吸附剂,铝系吸附剂可以选择性吸附LiCl,锰系吸附剂可以选择性吸附Li+,前者已经有了产业化的应用。在锂离子筛材料中,基于锂锰氧化物的锂离子筛(LMOs),由于有对 Li+的高选择性、高吸附量、无毒性以及低成本的特点而受到广泛关注[14-15]。 而影响离子筛吸附性能的因素也有很多,包括Mn3+的歧化反应、Jahn-Teller 效应[16]、离子筛的制备工艺、形态结构、晶粒尺寸、酸洗剂等,为了提高锰氧化物离子筛的提锂性能,众多的研究人员对其进行了研究,并提出了可以改善其性能的方法。
本文首先论述了锰系离子筛吸附剂的晶体结构, 解释了锰系吸附剂对锂离子选择性吸附的机理和吸附过程, 在此基础上综述了锰系吸附剂的制备方法, 最后从降低锰的溶解损失和提升吸附性能的角度对吸附剂改良的方法进行了归纳。
1 前驱体的结构与Li+脱出/嵌入机理
1.1 前驱体的结构
锰氧化物离子筛是目前锂离子筛的研究热点,常报道的有 LiMn2O4、Li1.33Mn1.67O4、Li1.6Mn1.6O4等,它们具有立方尖晶石型的晶体结构和三维网络通道,能够实现锂离子的吸、脱附过程。 以LiMn2O4为例,其前驱体的微观结构如图1 所示[17],其晶体结构属于Fd3m 群,氧原子呈面心立方堆积,锂占据四面体(8a)位,锰占据八面体(16d)位。 对于 Li1.33Mn1.67O4、Li1.6Mn1.6O4,锂占据四面体(8a)位和八面体(16d)位,锰占据八面体(16d)位。 Li+可以通过相邻四面体和八面体的间隙沿8a-16c-8a 的通道在三维网络结构中进行脱出和嵌入, 该八面体16c 空位被八面体16d 位置中的 6 个锰离子包围,如图 2 所示[18]。
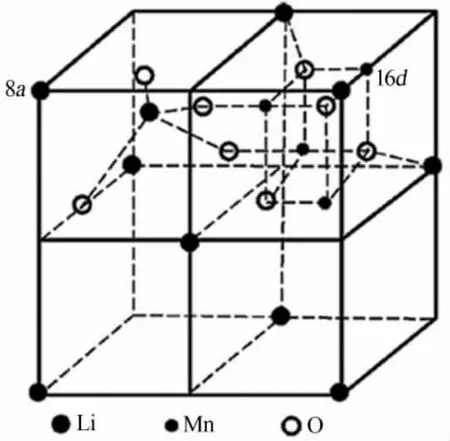
图1 LiMn2O4 的微观结构示意图[17]Fig.1 Schematic illustration of microstructure of LiMn2O4 [17]
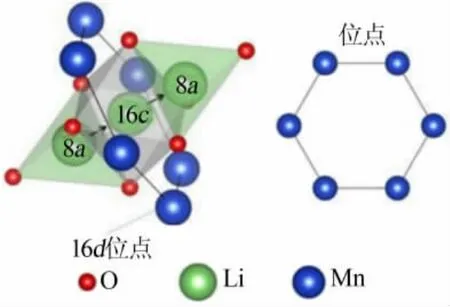
图2 锂扩散通道的示意图[18]Fig.2 Schematic illustration of the lithium diffusion channel [18]
1.2 Li+的脱出/嵌入机理
对于在吸附、 脱附过程中Li+的脱出和嵌入,目前主要有3 种机理进行解释:氧化还原机理、离子交换机理和复合机理。
1)氧化还原机理。 HUNTER[19]在制备 λ-MnO2中提出了氧化还原机理,在用酸洗离子筛前体LiMn2O4过程中,部分的Mn3+发生歧化反应,生成Mn2+和Mn4+,其中Mn4+在尖晶石结构中生成λ-MnO2,与此同时,Li+从内部脱出和部分的Mn2+溶于酸洗液中,此时锰的离子半径减小,从而晶胞参数也会变小。OOI 等[20]在研究Li+在λ-MnO2中的嵌入和脱出机理中认为其在吸附锂的过程中发生的反应为:
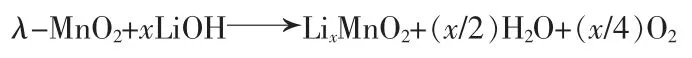
Mn4+会还原为Mn3+,Li+重新进入离子筛中,从而可进行下一步的循环吸附、脱附过程,但由于每次酸洗导致锰的溶损,其吸附性能会有所降低。
2)离子交换机理。 SATO 等[21]用 HNO3溶液酸洗 LiMn2O4后 生 成 的 λ-MnO2和 HMn2O4, 其 表 面锰离子的价态与LiMn2O4中锰离子的价态相同,认为在其表面发生了Li+和H+的离子交换反应:

对于观察到的 λ-MnO2表面的羟基,SATO 等[21]认为其与尖晶石氧化物结构中的空位8a 四面体位置有关,并且以与晶格羟基相同的形式存在。
3)复合机理。 虽然氧化还原机理和离子交换机理都可解释Li+在吸、脱附过程中部分嵌入和脱出的现象,但由于不同锂锰氧化物离子筛结构的差异性, 单个机理并不能完全地解释Li+的脱出/嵌入过程,因此FENG 等[22]提出了氧化还原反应和离子交换反应共同作用的复合机理。 在反应时,氧化还原型和离子交换型位点共同存在,且共用尖晶石的Mn—O 骨架,如图 3 所示[23]。 由图 3 可知,氧化还原型和离子交换型位点可以用锂锰氧化物的n(Li)/n(Mn)值和锰的平均氧化态来预测,每个位点的含量和比例取决于样品的制备条件。
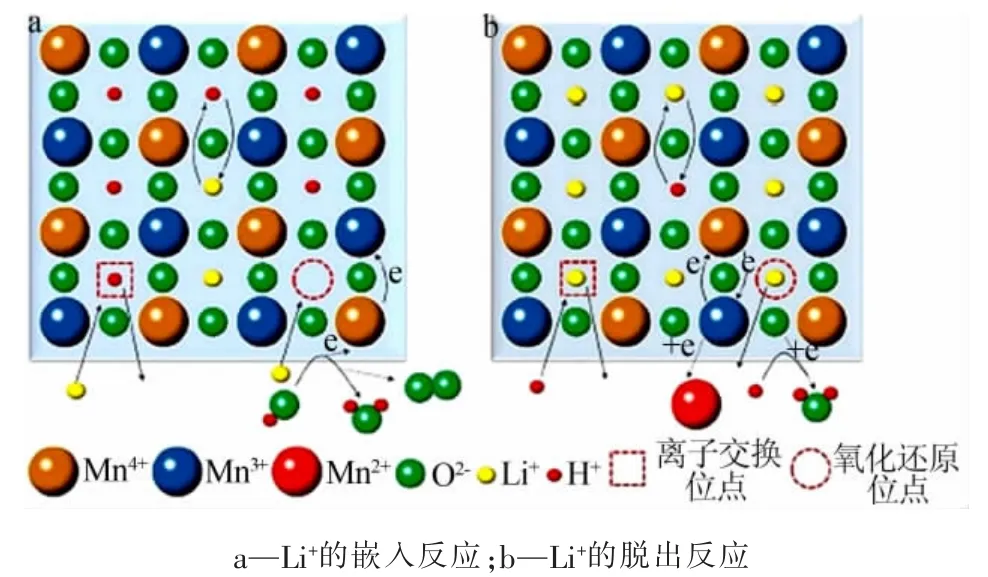
图3 Li+的嵌入、脱出反应复合机理示意图[23]Fig.3 Schematic illustration of Li+extraction reaction and insertion reaction by the composite mechanism[23]
2 锰氧化物离子筛前驱体的合成
要想得到离子筛, 首先需要制备出离子筛前驱体, 然后在不改变离子筛前驱体的晶体结构的前提下用酸进行处理,使H+取代Li+,将Li+抽取出来从而得到锂离子筛,其具有可靠的筛选和记忆效应,可以从多种共存的离子中吸附目标离子, 这种效应被称为“离子筛效应”。对于锂离子筛来说,只有锂离子可以进入空位, 因为它在所有金属离子中具有最小的离子半径。 离子筛的制备过程和离子筛效应如图4 所示[24]。 常用合成锂离子筛前驱体的方法有高温固相法、溶胶-凝胶法、水热合成法、微波烧结法、共沉降法[25]。 目前最常报道的锂离子筛的前驱体有LiMn2O4、Li1.33Mn1.67O4、Li1.6Mn1.6O4,这 3 种前驱体经酸洗后可分别得到离子筛 λ-MnO2、MnO2·0.31H2O 和MnO2·0.5H2O。

图4 离子筛制备过程和离子筛效应的示意图[24]Fig.4 Schematic illustration of the preparation process for ion-sieves and ion-sieve effect[24]
2.1 前驱体LiMn2O4 的合成
制备LiMn2O4最常用的方法为高温固相法和水热法,如FENG 等[26]通过长时间的水热反应制备出尖晶石氧化物并将其定义为 γ-MnO2;ZHANG 等[27]以 LiOH 和 Mn(NO3)2为锂源和锰源,用水热合成法制备出LiMn2O4,前驱体和最终的离子筛的形态为直径约为10~20 nm、长度约为几微米的纳米棒,这意味着软化学合成对形成具有均匀几何形状的LiMn2O4纳米结构是有效的。在用酸进行处理后,锂的脱出量达到5.51 mmol/g,最大锂离子平衡吸附量为酸处理去除锂量的 53.5%。 ÖZGÜR[28]利用超声喷雾热解法和固相合成法,以 LiNO3和 Mn(NO3)2·4H2O 为锂、锰源制备出吸附性能较高的LiMn2O4。 其中,固相反应制得的粉末具有典型的LiMn2O4形貌: 针状结构、边缘尖锐、表面粗糙,而喷雾热解制备的LiMn2O4由于较小球形颗粒的团聚粉末呈球形,如图5 所示。

图5 LiMn2O4 的 SEM 照片[28]Fig.5 SEM images of LiMn2O4[28]
SUN 等[29]用LiOH 和 MnO2在高温煅烧的条件下制得 LiMn2O4。 JI[30]用高温固相法合成 LiMn2O4,以Na2S2O8为洗脱剂,合成的前驱体和离子筛均为尖晶石结构,且无团聚现象。锂的提取率达到93%以上,由于SO42-自由基的抑制作用,锰几乎没有损失。
2.2 前驱体 Li4Mn5O12(Li1.33Mn1.67O4)的合成
Li4Mn5O12是锰氧化物锂离子筛前驱体之一,最早由 THACKERAY[31]合成,其合成方法有固相合成法、水热合成法及溶胶-凝胶合成法等。 如TAKADA 等[32]用溶胶-凝胶法,通过在 O2气氛下将LiOAc 和 Mn(NO3)2的共晶混合物加热至 700 ℃制备出结晶良好的 Li4Mn5O12。 TANAKA 等[33]用固相合成法制备出平均粒径为 100 nm 的Li4Mn5O12,且在600 ℃时形成了结晶良好的纯尖晶石相。 XIAO 等[34]、LIU 等[35]、GAO 等[36]用两步煅烧法合成了 Li4Mn5O12,颗粒的直径在100 nm 左右,且有团聚现象。 LIU等[35]以乙酸为 Li+洗脱剂,前驱体在 0.5 mol/L 乙酸中进行洗脱, 锰的溶解率仅为0.01%, 且在pH 为2.0 左右时能够实现Li+从Li4Mn5O12中的解吸过程。
2.3 前驱体Li1.6Mn1.6O4 的合成
目前,H1.6Mn1.6O4被认为是吸附量最高、 稳定性最好的锰氧化物离子筛, 其前驱体Li1.6Mn1.6O4的理论离子交换能力是所有锂锰氧化物中最高的, 可达10.5 mmol/g[37],因此,前驱体 Li1.6Mn1.6O4的制备已成为研究热点。传统的合成方法有固相合成法、液相合成法及溶胶凝胶法。 大多数研究者采用水热法先合成 LiMnO2, 后经过高温烧结合成 Li1.6Mn1.6O4。 如CHITRAKAR 等[38]用 γ-MnOOH 和 LiOH 为原料 先经水热法合成LiMnO2,后在400 ℃空气氛围下烧结4 h 合成 Li1.6Mn1.6O4,其吸附量高达 40 mg/g,用酸处理后,锂的脱附率可达99%,锰的溶损率仅为2%。SHI 等[39]以 LiOH 和 Mn2O3为锂锰源,通过水热合成和高温烧结的方法制备出Li1.6Mn1.6O4, 研究发现,在410 ℃时,锰酸锂中的所有Mn3+可被氧化成Mn4+,可获得Li1.6Mn1.6O4的纯产物,当焙烧温度为500 ℃时,其产物为立方尖晶石结构的Li1.6Mn1.6O4和单斜晶体Li2MnO3组成的混合物。 经过10 次循环后其吸附量仍能高于21 mg/g, 最后得到的碳酸锂的纯度在99.5%以上。
GAO 等[36]在研究锰酸锂中锂的吸脱附机理时,以LiOH·H2O、KMnO4和Mn(NO3)·24H2O 为原料,通过先水热、后烧结的方法合成Li1.6Mn1.6O4,同时通过实验和理论分析,阐明了锂离子筛的解吸机理,包括不同位置的锂离子扩散通道。还有的研究者使用相同的方法,通过替换锂、锰源合成Li1.6Mn1.6O4,其各自的吸附量有较大的差异,是由于实际的盐湖卤水或海水与模拟的富锂液相比存在着较多的杂质和干扰离子,因而锂离子筛在海水和卤水中的吸附量相对较低[40]。
LI 等[41]通过水热合成、固相反应合成等方法制备了棒状、 球状、 花状和三维多孔结构的锂离子筛(见图6)。 结果表明,不同形貌的锂离子筛在比表面积、吸附选择性和结构稳定性方面存在很大差异。三维多孔结构的离子筛具有较高的吸附容量和吸附速率,但锰的溶解损失较大,其吸附稳定性较差,而球状的离子筛具有均匀的粒径和较好的分散性, 因此对卤水中的锂离子具有良好的吸附性能。

图6 离子筛的 SEM 照片[41]Fig.6 SEM images of ion-sieves[41]
除传统的合成方法外, 研究人员也在寻求一些新的方法,以避免合成方法繁杂、合成产物不均匀、合成所需温度较高等缺点,如 CHEN 等[42]使用微波辐射和高温煅烧的方法合成Li1.6Mn1.6O4,其在材料合成方面具有很好的应用前景。
3 锰氧化物离子筛的改性
3.1 掺杂改性
虽然锰氧化物离子筛对锂具有极高的选择性和较高的吸附容量,但在工业化应用过程中,锰的溶损和Jahn-Teller 效应仍是限制其发展的两个重要因素,为了减小锰的溶损,抑制Jahn-Teller 效应,众多研究者对此进行了研究。其结果表明,在合成锰氧化物离子筛过程中掺杂阴离子或金属阳离子可有效降低锰的溶损,提高锰氧化物尖晶石结构的稳定性。
3.1.1 阴离子掺杂
锰氧化物离子筛前驱体中的氧被其他阴离子部分取代可形成阴离子掺杂的尖晶石型锂锰氧化物。由于离子半径由大到小依次为S2-、O2-、F-,氟离子更容易进入晶格,而硫的化学性质与氧相似,因此可选择 F-、S2-进行阴离子掺杂,如 QIAN 等[43]在 350 ℃条件下通过添加不同比例的 NH4F 和(NH4)2S 与LiMnO2混合后煅烧24 h,制备出氟和硫掺杂的尖晶石锂锰氧化物的离子筛前体(LMO-R,R=F、S),掺杂后的离子筛对Li+的吸附量(33.4 mg/g 和27.9 mg/g)高于未掺杂的离子筛(26.1 mg/g)。 离子筛性能的提高依赖于氟和硫在32e 位上对氧的取代。 尽管阴离子掺杂可有效提高离子筛对Li+的吸附量,然而在一定程度上也会加剧Jahn-Teller 效应,制约其工业化应用, 因此国内外学者对于阴离子掺杂离子筛的研究较少,大多数的研究集中于金属阳离子的掺杂。
3.1.2 金属阳离子掺杂
锰氧化物离子筛前体中八面体空隙的锰离子被其他金属阳离子部分取代时, 可形成金属阳离子掺杂的尖晶石型锂锰氧化物离子筛。 掺杂阳离子的选择因素有[44]:1)选择与 Li+或 Mn3+半径相近的阳离子, 掺杂前后锰氧化物尖晶石结构不会出现明显的膨胀或收缩;2)选择低价态的阳离子进行掺杂可以提高锰离子的平均价态, 从而抑制Mn3+的歧化反应;3)选择与氧形成的键能高于Mn—O 键的键能的阳离子,可提高锰氧化物尖晶石结构的稳定性。对于所掺杂的阳离子,常报道的有 Al、Mg、Co、Ni、Fe 等,其微量掺杂进入锂离子筛前驱体中不会改变原有的尖晶石结构。
Al3+的半径(0.067 nm)比 Mn3+的半径(0.079 nm)小,掺杂Al 进入尖晶石锰氧化物的晶格中会导致晶格收缩,原子间的作用力增大,同时铝离子部分取代锰离子形成的Al—O 的键能大于Mn—O 的键能,使晶体结构更加稳定。 如 CHEN 等[45]通过水热法合成了Al 掺杂的LiMn2O4,掺杂后锂离子筛的结构更稳定,5 次循环后仍能保持较高的吸附容量(19.5 mg/g),同时锰的损失极低。许乃才等[46]研究了固相掺杂氯化镧和硝酸铝对锂离子吸附性能的影响, 结果表明Al3+掺杂的离子筛产生的活性吸附位点最多,La3+掺杂的离子筛次之, 而不掺杂的锰氧化物拥有的吸附位点最少。 ZHANG 等[47]通过溶胶凝胶法和固相法合成Al 掺杂的Li1.6Mn1.6O4,Al 均匀地分布于离子筛晶体结构中,掺杂Al 的离子筛的吸附容量(32.6 mg/g)高于未掺杂的离子筛(27.6 mg/g),锰的溶解率从2.06%下降到1.96%, 同时循环稳定性也有所增强。
由于 Mg2+的半径(0.086 nm)与 Li+的半径(0.088 nm)相近,且 Mg2+的水合能(-1 862 kJ/mol)远大于Li+的水合能(-510.4 kJ/mol),因而镁掺杂的锂离子筛能够对Li+表现出特殊的选择性[48]。 如TIAN等[14]采用软化学方法合成了具有镁掺杂的锂锰氧化物前驱体LiMg0.56Mn1.5O4,酸洗后得到锂离子筛H1.51Li0.08Mg0.24Mn1.49O4,当模拟液的pH 为 12、初始质量浓度为200 mg/L 时, 离子筛对锂的吸附量达到37.4 mg/g,4 次循环后,其饱和吸附量仍不低于初始吸附量的 95%。 CHITRAKAR 等[49]采用溶胶凝胶法和固相法合成镁掺杂的离子筛前驱体LiMgzMn2-zO4,掺杂前后吸附量由40 mg/g 增加到43.7 mg/g,锰的溶损率由5.8%降低至2.5%, 很大程度上改善了锂离子筛提取锂的性能。 钱方仁等[50]通过水热法和固相合成法制备出了 Mg2+掺杂的 Li1.6Mn1.6O4(LMMO),循环吸脱附过程中,Mg2+替换体相16d 位点的 Li+,由于Mg—O 键强于Li—O 键,掺杂后能够增强材料的结构稳定性,同时Mg2+掺杂后,降低了16d 位点附近的电负性,提高了Mn 的平均价态,因而使Mn 的溶损率降低。
Cr3+的半径(0.075 5 nm)与 Mn3+的半径(0.079 nm)相近,同时 Cr—O 键能(1 029 kJ/mol)比 Mn—O 键能(946 kJ/mol)大,选择Cr3+微量掺杂进入锂离子筛前驱体结构中,可提高锰的平均化合价,抑制Jahn-Teller 效应,增强晶体结构的稳定性。 如 CAO 等[51]通过水热法和固相法合成了Cr 掺杂的Li1.6Mn1.6O4,与未掺杂的锂离子筛相比,Cr 掺杂的离子筛提高了锂的吸附容量, 对离子筛的结构稳定性有明显的改善作用。
还有其他研究人员通过掺杂Fe3+,合成锰基锂离子筛前驱体,如LI 等[52]通过水热法和固相合成的方法制备出了Fe 掺杂的离子筛前驱体Li1.6Mn1.6-xFexO4,Fe3+被引入锂离子筛晶格,且当 x 为 0.032 时,锂的吸附容量可达到35.29 mg/g,锰的溶损率为1.66% 。 CHITRAKAR 等[53]通 过 固 相 反 应 合 成Li1.33FexMn1.67-xO4,吸附量较未掺杂的离子筛有较大提高。 在酸洗过程中锰和铁的损失极小。 WANG 等[54]和ZHAO 等[55]也研究了铁掺杂的离子筛,结果表明在酸洗过程中锰的损失极小, 有较好的吸附性能。QIAN 等[56]将 FeCl3、CoCl2和 NiCl2分别与 LiMnO2混合后在 350 ℃的条件下煅烧 24 h,制备出 Fe、Co、Ni掺杂的锂离子筛前驱体(LMO-R,R=Fe,Co,Ni),并研究了掺杂前后离子筛的性能,其中HMO-Fe 的吸附量(35.3 mg/g)、HMO-Co 的吸附量(35.4 mg/g)与HMO-Ni 的吸附量(33.6 mg/g)均高于未掺杂的锂离子筛 HMO 的吸附量(32.3 mg/g),但掺杂 Ni 的锂离子筛有较大的锰损失。
还有的研究人员研究了Na+、K+、Zn2+掺杂锰基离子筛,如 FENG 等[57]以 LiOH、Mn(NO3)2和 Zn(NO3)2为原料, 通过共沉淀法制备出锌掺杂的离子筛前驱体LiZn0.5Mn1.5O4。QIAN 等[58]通过在 Li1.6Mn1.6O4(LMO)表面微量掺杂Na+, 建立了一种提高锂离子筛结构稳定性和吸附性能的有效方法,HMO-Na 对锂的吸附量(33.9 mg/g)高于未掺杂的 HMO(33.5 mg/g),LMO-Na 中锰的溶解率(4.4%)低于 LMO(5.4%),离子筛性能的提高依赖于Na+对16d 位置Li+的取代。QIAN 等[59]还通过掺杂 KCl 制备出 K+掺杂的离子筛前驱体(Li1-xKx)1.6Mn1.6O4,用于提高锂离子筛结构的稳定性,当x 为2%时,其性能最好,锂的吸附量可达到31.6 mg/g,锰的溶解率也从5.4%降至4.0%。
以上以改善锰基离子筛性能为目的的阴、 阳离子掺杂,可有效地提高锂的吸附容量,降低锰的溶解损失,改善锂离子筛的性能,但不可否认的是其仍未能从根本上解决锰的溶解问题, 在循环过程中仍旧有较大的损失,同时由于掺杂引入了其他元素,为后续制备高纯度锂盐也增加了一定的难度。 如何更大程度地改善离子筛的性能、 提高其稳定性仍是今后需要解决的难题之一。
3.2 包覆改性
改善离子筛的性能除了在其合成过程中进行掺杂, 还可以在锂离子筛前驱体的表面进行金属氧化物的包覆, 以降低离子筛在酸洗过程中对锰溶解的影响,如 OHASHI 等[60]采用浸渍法在可控制酸碱度的溶液中合成了一系列表面改性的锂离子吸附剂,将Al 和Ni 的氧化物包覆在Li1.33Mn1.67O4的表面,然后对其进行热处理, 金属氧化物层在基体表面形成并与基体结合。 与未包覆的Li1.33Mn1.67O4相比,金属氧化物包覆后的离子筛显示出较低的锰的溶解率和较高的锂的吸附量。
王豪等[61]采用水热法制备前驱体 Li1.6Mn1.6O4,并用液相沉淀法在其表面包覆ZrO2,酸洗后形成包覆ZrO2的锂离子筛H1.6Mn1.6O4,且当ZrO2包覆量为3%(质量分数)、焙烧温度为450 ℃时,可在前驱体表面形成厚度约为15 nm 的ZrO2包覆层,锰的溶损率从3.14%下降到2.65%,锂离子筛对锂的吸附容量保持在 29.4 mg/g,高于未包覆的锂离子筛(22.9 mg/g)。包覆ZrO2改善了锂离子筛的结构稳定性,提高了吸附容量。
杨喜云等[62]采用溶胶-凝胶法在 LiMnO2表面包覆TiO2,再经焙烧得到包覆TiO2的离子筛前驱体Li1.6Mn1.6O4,当包覆量为3%(质量分数)时,离子筛在盐湖卤水中的吸附容量为24.3 mg/g, 循环5 次后,锰溶损率仅为0.4%(质量分数)。 离子筛经TiO2包覆改性后,锰溶损显著降低,吸附容量无显著变化,循环稳定性明显提高。
3.3 其他方式
由于离子筛粉体在吸脱附过程中容易造成较大损失, 锂离子筛的成型技术对于从海水或卤水中回收锂具有重要意义。 在工业化应用中通常将离子筛粉体分散在聚氯乙烯(PVC)、聚氨酯(PU)、聚乙烯醇(PVA)和聚丙烯腈(PAN)等聚合物中[63-66]。 离子筛粉体和聚合物的混合物可以用于造粒、 成膜或制成纳米纤维等,以获得复合材料。
JIA 等[67]以 亲 水 性 聚 丙 烯 腈 (PAN) 和Li1.6Mn1.6O4(LMO)为黏结剂和前驱体,通过相转化法制备了不同类型的H1.6Mn1.6O4复合吸附剂, 包括颗粒(HMO/PAN-G)、纳米纤维平板膜(HMO/PAN-NM)、平板膜(HMO/PAN-FM)和中空纤维膜(HMO/PANHFM)。CHUNG 等[68]将聚丙烯腈(PAN)和 Li1.6Mn1.6O4溶解在二甲基甲酰胺(DMF)中,搅拌均匀后使用静电纺丝设备制成复合纳米纤维材料, 可以成功地用于海水提取锂离子的多次循环过程。 ZHU 等[69]以尖晶石型锂锰氧化物粉末Li1.6Mn1.6O4为前驱体、 聚氯乙烯(PVC)为黏结剂、二甲基乙酰胺(DMAC)为溶剂, 采用溶剂交换法制备出可多次使用的复合材料PVC-Li1.6Mn1.6O4。 QI 等[70]、QIU 等[71]和 SUN 等[72]利用不同的黏结剂分别制备出PVDF-Li1.6Mn1.6O4多孔膜、PVA/CAM-Li4Mn5O12纤维素膜和大孔聚合物离子膜,用于分离卤水中的锂。
LAI 等[73]和 HONG 等[9]分别采用环氧树脂和氧化铝凝胶制备出了具有优异吸附性能的柱状颗粒吸附剂(EP/HMO 复合材料)和多孔的柱状HMO/Al2O3复合材料(见图 7)。 ZHAO 等[74]采用溶胶-凝胶法成功制备了吡咯/氧化铝的三维纳米结构材料(PPy/Al2O3/LMO),其能有选择性地从卤水中分离锂离子,并具有长期稳定性。

图7 HMO/Al2O3 复合材料[9]Fig.7 HMO/Al2O3 composite materials[9]
上述所制备的复合材料可大大地提高其循环使用性能、降低离子筛基体的损失,但同时也会降低对Li+的吸附能力和动力学性能[75-77]。 其原因包括质量传输限制、离子筛复合物吸附位点的聚合物堵塞以及由于聚合物基质的疏水性导致的进料亲和力降低[69,78]。
4 结论及展望
在过去的几十年里, 随着锂离子电池的快速发展和广泛应用,全球对锂资源的需求显著增加。由于盐湖卤水中存在着大量锂资源, 从盐湖卤水或海水中提取锂将是获取锂资源最重要的途径, 而锰基锂离子筛又具有对锂离子的高选择性、化学稳定性、无毒性以及低成本的特点, 因而将会是后续提锂材料研究的重要方向。
目前对锂离子筛的改性主要集中于掺杂、 表面包覆以及合成复合材料这三方面, 其在一定程度上可提高离子筛的性能, 但仍未能根本解决离子筛在酸洗过程中锰的溶解问题, 大多数所报道的锂离子筛也只局限于实验室中的低数量级循环, 无法满足工业化大生产中高数量级循环的要求,因而在锂离子筛改性方面的研究仍然有待深入, 需要继续探索结构稳定的离子筛材料,以获得提取能力和循环稳定性之间的良好平衡。 为了提高工业应用中锂离子筛的性能,应从高选择性、高稳定性、低成本和环境友好性的角度对离子筛材料进行进一步的开发和优化。
猜你喜欢
中国宝玉石(2022年2期)2022-04-25
有色金属科学与工程(2022年1期)2022-03-12
陶瓷学报(2021年4期)2021-10-14
陶瓷学报(2021年1期)2021-04-13
燃料化学学报(2020年3期)2020-05-07
大连工业大学学报(2020年1期)2020-01-17
武汉工程大学学报(2019年5期)2019-11-02
汽车生活(2017年4期)2017-04-26
山东工业技术(2016年15期)2016-12-01
中学化学(2015年9期)2016-04-14
