
基于德尔菲法的《临床试验生物样本伦理管理指南》研究*
2022-02-08 08:21徐文华汪秀琴
医学与哲学 2022年18期
徐文华 黄 旭 汪秀琴
生物样本是药物研发与生物技术的重要物质和战略资源,在发病机制探索、生物标志物开发、新药研究测试、疾病精准诊疗等方面意义重大。人类生物样本以造福人类健康为宗旨,但是在实际应用中却引发了知情同意、隐私保护、样本商业化、捐赠者权益、利益冲突、样本共享等诸多伦理问题[1]。以全球合作为特征的生物样本库3.0 版时代还将引发种族伦理和国家安全等更多新问题[2]。
在江苏省药品监督管理局《2020 年度药品监管科学科研计划课题》支持下,本项目组立足前期相关研究基础[3-6],借鉴发达国家经验做法,结合我国临床试验生物样本具体实际,通过两轮德尔菲法进行权威专家论证,形成国内首个《临床试验生物样本伦理管理指南》,旨在规范临床试验中生物样本的管理与伦理审查,促进药物与器械临床试验的健康发展。现将指南制定过程和全文发布如下,供行业参考借鉴。
1 资料与方法
1.1 文献检索与研究
检索中英文数据库20 个,包括PubMed、Web of Science、Cochrane Library、万方数字化期刊、中国知网、美国国立临床诊疗指南数据库、国际指南协作网、英国国家卫生与临床优化研究所指南检索网站等。检索日期为建库至2021 年3 月,语言为英文和中文。中文检索词关键词包括:生物样本、生物技术、伦理、伦理委员会、指南、临床试验。英文检索词包括:biobank、biological technology、ethic、ethics review committee and/or institutional review board、guideline、clinical trials。同时手工检索收集欧美发达国家、国际组织以及中国发布的生物样本库管理法规与伦理指南[7-15]。对检索的重要文献以及国际国内指南规范进行研究,结合《江苏省重大疾病生物样本库伦理指南》,初步拟定《临床试验生物样本伦理管理指南》条目与内容。
1.2 形成征求意见稿
由课题组成员以及多名国内知名伦理专家组成课题专家组,通过会议讨论方式对拟定的《临床试验生物样本伦理管理指南》条目与内容进行充分讨论、分析与建议,然后形成《临床试验生物样本伦理管理指南征求意见稿》。
1.3 建立咨询工作组,编制专家咨询表
课题负责人组织课题组成员完成以下工作。
(1)确定咨询专家组:采用同行推荐方式从科研伦理学、生物样本库、生物技术领域筛选国内权威专家。专家领域包括医院管理、伦理学、临床医生、生物样本管理、创新生物技术从业者以及法学专家。咨询专家标准:①科研伦理、生物样本库、生物技术、临床医学或者法学领域工作者;②至少5 年相关领域工作经验;③本行业知名专家和权威专家;④对本研究有兴趣且愿意参加。咨询专家组的基本情况见表1。
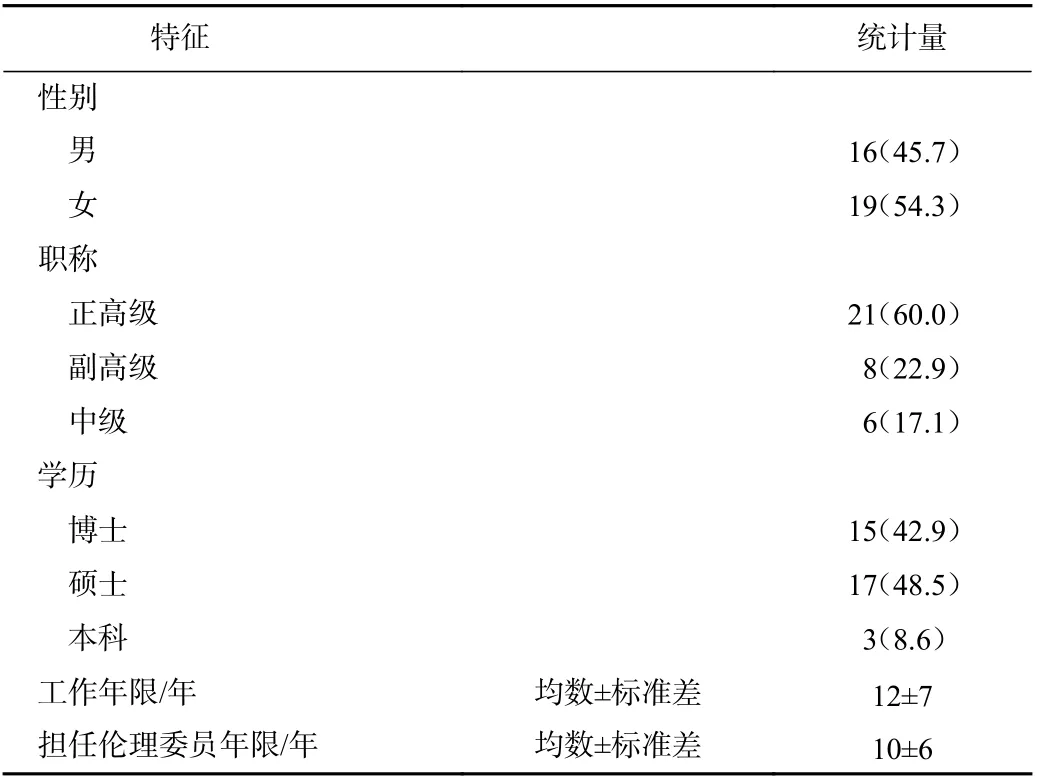
表1 咨询专家基本特征(n=35) 单位:n(%)
(2)编制专家咨询表:专家咨询表包括4 部分:第一部分:专家基本信息。第二部分:《临床试验生物样本伦理管理指南》条目与内容。对每一条目按照1 分~5 分进行重要性赋值(Likert 评分法:1 分为完全不重要,2 分为不太重要,3 分为一般,4 分为比较重要,5 分为非常重要)。第三部分:专家建议指南需补充的条目与内容。第四部分:专家对调查内容熟悉程度进行自我评价。
1.4 开展两轮德尔菲法专家组咨询论证
通过电子邮件发送专家咨询问卷,或者以电话/面对面访谈形式收集专家意见。正式调查前请5 名专家进行预调查,测试调查步骤合理性与问卷方案完整性。开展德尔菲法专家咨询,对指南条目与内容进行权威论证,根据第一轮专家意见调整后的条目进行第二轮咨询。
1.5 条目筛选方法
通过两轮德尔菲法国内外专家组调研问卷进行咨询表数据处理与结果分析,参考重要性评分、变异系数等,召开课题专家共识会议,经专家讨论决定筛选条目。
1.6 统计学分析
使用Excel 2013 软件建立专家咨询组结果数据库,使用SPSS 26.0 软件进行统计分析。计量资料以均数±标准差表示,计数资料以频数、百分比表示。咨询专家的积极性系数用咨询问卷回收率和提出具体文字修改建议的专家参与率表示。用Kendall 协调系数表示咨询专家意见的协调程度,协调系数越接近1 表示专家对指南结构认同的一致程度越高;对协调系数进行χ2检验,检验水准α=0. 01。咨询专家的意见集中程度反映了专家对指南条目的总体意见,通过均数、标准差、变异系数和满分率表示。咨询专家对条目的评价通过条目评分体现,使用平均数、标准差、变异系数和满分率描述条目评分的分布。
2 德尔菲法分析结果
2.1 专家咨询积极性
本课题共进行了两轮德尔菲法专家咨询。两轮咨询均发放专家意见咨询问卷38 份,均回收35 份,回收率为92.10%。对指南条目提出具体文字性修改意见的专家分别为35 名和28 名,专家参与率为92. 10%和73.68% 。
2.2 专家咨询意见协调程度
两轮德尔菲法专家咨询,协调系数分别为0.236,0.375,均P<0.001。具体见表2。

表2 两轮德尔菲法专家意见协调系数
2.3 指南条目筛选
两轮德尔菲法专家咨询之后,专家意见趋于一致,故不再进行第三轮咨询。根据条目筛选方法,最终确定《临床试验生物样本伦理管理指南》由26 个条目构成,见表3。

表3 第二轮专家咨询指标重要性评价结果
通过两轮德尔菲法专家组调研问卷进行调查表数据处理与结果分析,之后召开课题组专家共识会议,撰写指南初稿,然后组织权威专家内审会讨论,最终形成《临床试验生物样本伦理管理指南》终稿,现将指南全文发布如下。
第一章 总则
第一条 【宗旨目的】为规范临床试验中人类生物样本及相关信息(下称生物样本)的管理与伦理审查,保障样本提供者的权利和福利,促进生物医学研究健康发展,增进人民健康福祉,特制定本指南。
第二条 【法规指南】临床试验中涉及生物样本的管理与伦理审查应当符合《赫尔辛基宣言》《涉及人的健康相关研究国际伦理指南》原则与伦理要求,遵守《中华人民共和国生物安全法》《中华人民共和国数据安全法》《中华人民共和国个人信息保护法》《中华人民共和国人类遗传资源管理条例》《涉及人的生物医学研究伦理审查办法》《药物临床试验质量管理规范》《医疗器械临床试验质量管理规范》等法律规范,遵照尊重、有利、不伤害、公正的伦理原则,倡导透明、共享和负责任的研究利用[7]。
第三条 【适用范围】本指南适用于在医疗卫生机构中开展、由申办者发起的涉及生物样本的药物和医疗器械(含体外诊断试剂)临床试验。研究者发起的临床研究可参考本指南。
第四条 【共治责任】临床试验机构、研究者、伦理委员会、申办方、检测机构和样本保藏机构等均有责任保护生物样本提供者的权益和安全,应当按照法律法规和伦理要求做好生物样本的采集、保藏、利用和销毁等工作。禁止利用样本开展危及公众健康、危害生物安全的研究开发活动。
第二章 管理体系
第五条 【责任主体】 临床试验机构应当遵守国家法律法规和伦理规范,建立生物样本管理体系,制定生物样本全流程管理制度,指定专门部门负责生物样本的管理,制定样本采集、保藏、利用、对外提供、销毁等环节操作规程,保护样本提供者的权益和安全,做好样本的生物安全保障工作[8]。伦理委员会应对每一项涉及生物样本的试验方案进行审查。研究者应在获得样本提供者的知情同意后采集生物样本。
第六条 【制度规范】临床试验机构应建立制度规范保障生物样本用于具有科学性与社会价值的研究,不得买卖人类生物样本。确保获得样本提供者的同意,保障样本提供者撤回同意的权利,保障样本提供者隐私和可识别数据的机密性,建立生物样本库的质量管理体系,记录生物样本的采集数量、转移和使用情况、剩余样本去向等,建立向样本提供者反馈研究结果的机制。
第七条 【共享利用】生物样本的重要价值在于样本利用与数据共享。样本共享利用应该重点考虑科学与社会价值,以及是否合法合规和符合伦理要求。样本共享利用应委托给具有法人资格的有资质的单位。
第八条 【转移协议】临床试验机构应与样本利用与管理方签署生物样本转移协议。协议应约定样本转移种类、形式、数量、用途等,明确各方的权利、义务和责任,知识产权、研究成果分配与数据结果返还等问题。涉及样本数据的国际合作与出境必须获得相关部门批准。
第九条 【成果回馈】利用生物样本开展研究所取得的成果应进行共享,包括数据反馈、数据平台共享、知识产权分享等形式。利用生物样本产生商业利益的时候,应考虑建立回馈样本提供者所属人群或特定群体的机制。
第十条 【利益冲突】临床试验中涉及生物样本的采集、保藏、利用、对外提供等工作倡导公开透明原则。如涉及相关利益冲突,应向样本提供者进行告知。
第十一条【结果反馈】利用生物样本开展研究可能对样本提供者及其亲属产生具有重要意义的研究结果,研究者应充分尊重样本提供者的个人意愿,慎重考虑是否需要告知以及如何告知。一般而言,研究结果反馈应遵循分析有效性、临床重要性以及反馈可行性的原则。
第三章 知情同意
第十二条 【知情同意】临床试验中涉及生物样本采集、保藏与利用应获得样本提供者的知情同意。应告知样本提供者有随时撤回同意的权利及撤回方式。主研究以外的生物样本采集,建议提供一份单独的生物样本采集知情同意书,或者具有醒目标识的单独段落进行告知并分别同意,不得将此类生物样本采集作为参与主研究的条件。
第十三条 【信息告知】临床试验中涉及生物样本的采集,应当获得样本提供者事先、自愿和明确的同意。应向样本提供者说明可以在任何时候撤回其同意的意见,而且不会因此受到伤害或歧视。告知信息应与试验方案中相关内容一致,一般应包括:
(一)生物样本的采集、储存、保藏、利用与销毁:采集目的,样本种类,采集方法、部位与数量,采集数据范围,保藏条件、保藏期限与销毁规定;如适用,应告知将来的生物样本研究领域与利用范围;
(二)采集、储存、保藏和利用生物样本的机构/相关合作方;
(三)对样本提供者可能造成的风险及对他人的影响;承诺样本采集不会影响临床诊断和治疗,告知是否会因样本采集而扩大手术切除范围或增加采集量、采集次数和采集种类,如需额外采集,应明确告知;
(四)研究结果可能给其他患者和社会带来的益处;
(五)隐私与可识别数据的保护措施;
(六)自愿参与和撤回知情同意的权利,撤回后生物样本和相关数据的处理方式,同时说明撤回知情同意的局限性;
(七)利用生物样本获得的研究结果是否告知样本提供者以及告知方式;
(八)样本提供者的权益,如没有,则明确说明;提供样本可能获得的补偿;
(九)生物样本或数据是否会共享及共享范围。
第十四条 【签署知情同意】应以能够理解的语言告知必要和完整的信息,在样本提供者充分理解、自愿同意的情况下签署知情同意书。
第十五条 【弱势受试者】对于涉及弱势受试者的生物样本采集,应当慎重考量科学价值与样本提供者权益,必要时须获得监护人同意。
第十六条 【泛知情同意】建议慎重采用泛知情同意采集与保藏临床试验生物样本。如采集研究剩余的或者临床诊疗剩余的生物样本用于目的尚不明确的未来研究,研究者与申办者等应承诺,未来研究开展之前将获得伦理委员会审查批准后再实施。
第十七条 【再次知情同意】如果利用生物样本开展超出之前知情同意告知范围的研究时,以及研究过程中发生实质性变化,应提交伦理委员会审查,一般情况下应获得样本提供者的再次知情同意。
第四章 伦理审查
第十八条 【体系审查】开展临床试验的医疗卫生机构应将生物样本管理体系文件提交伦理委员会审查,包括:
(一)临床试验生物样本管理制度,包括生物样本采集、转运、储存、保藏、销毁及质量管理操作规程;
(二)生物样本库设施条件,或者生物样本采集、储存、保藏的场地与设备条件;
(三)生物样本管理人员资质;
(四)泛知情同意书;
(五)隐私与保密制度、数据安全相关规定;(六)生物样本合作转移协议模板;
(七)生物安全管理规定;
(八)其他材料。
第十九条 【项目审查】涉及生物样本的临床试验应向伦理委员会提交材料或者补充说明,包括:
(一)伦理审查申请表:说明项目概况、样本提供者人群、获益与风险、样本使用、个人信息保密、数据保护、结果披露、退出研究、联系方式等;
(二)研究方案:应说明采集样本的目的、对象、方法,样本采集量的估算方法,明确样本类型、样本采集量、样本数量等信息;
(三)知情同意书;
(四)样本合作利用:提供申办者、检测机构和样本保藏机构信息,及其遵守法规与伦理要求的承诺书;
(五)其他材料。
第二十条 【审查要素】伦理委员会应对临床试验中涉及生物样本的伦理问题进行审查与考虑,审查要素包括:
(一)生物样本采集计划是否合适,样本采集的必要性、科学性和合理性,考虑研究目的、样本采集者、采集方式、采集时限、样本例数、样本类型、采集数量、采集量、样本用途、采集数据等;
(二)研究人员资质与经验是否符合要求;
(三)样本提供者可能的风险、获益与保护措施;
(四)在知情同意过程中,向样本提供者提供的告知信息是否完整易懂,获得知情同意的方法是否适当,样本提供者是否可以撤回同意及撤回同意后样本数据的处理;是否将采集生物样本知情同意书的签署作为参加主研究的必要条件;
(五)样本提供者隐私与可识别数据保护措施是否合适;
(六)是否提供补偿,补偿与相关费用是否合适;
(七)样本共享申请人(申办者、检测机构和样本保藏机构)的法人资格与相关资质是否符合要求,是否涉及利益冲突问题,结果回馈与样本销毁计划是否合适;
(八)样本保藏机构与委托样本储存机构应建立合格的生物样本库,具有相应场所、设施设备、质量管理与样本库人员,具有严格的隐私保护与数据保密规定;
(九)涉及致病性病原微生物的样本采集、保藏、转运和研究,应符合生物安全管理规范。
第五章 隐私和保密
第二十一条 【各方责任】涉及生物样本采集、保藏、利用、对外提供、销毁等环节的所有相关机构及工作人员(以下统称“相关方”),包括临床试验机构、申办者、合同研究组织/临床试验现场管理组织、检测机构、样本储存/保藏机构等均有责任保证样本提供者隐私保护与可识别数据机密性,避免信息泄露可能给样本提供者带来的伤害、污名和困扰[9]。
第二十二条 【保密制度】相关方应制定隐私保护和数据保密制度,包括一旦出现隐私和可识别数据泄露时的应急预案。应当对工作人员进行培训,并签署保密承诺书。
第二十三条 【隐私保护】相关方应采取严格措施保护样本提供者的个人隐私信息,包括姓名、身份证件号码、出生日期、电话号码、社交媒体账号、住址、住院号/门诊号、医保卡号、生物识别信息(如面部照片)、遗传信息等可识别个人身份信息。
第二十四条 【数据保密】管理样本和数据的信息化系统应设置完善的权限管理,访问过程应留痕并可以溯源,未获得授权人员不能访问;纸质材料应上锁并由专人保存;存储在系统中的数据必须通过匿名或编码、加密、去链接等方式加以保护;需要将样本提供者个人信息与样本数据进行关联分析时,应建立严格的审核与管控程序;未经样本提供者同意,个人相关疾病信息不能透露给他人。
第二十五条 【样本转移】进行样本转移时应采取保密措施,向申办者提供的数据只能以匿名或编码的方式提供,不得向样本接收方转移样本提供者姓名、身份证号码、出生日期、电话号码等可识别个人身份信息。
第二十六条 【数据安全】相关方应符合国家网络安全等级保护制度要求,具备数据安全管理制度和相关设施设备,包括服务器、电脑终端、防火墙等保障网络和信息化安全,并建立数据安全技术与数据备份机制来保障数据安全。
术语及释义
人类生物样本[10]:指从人体获得或衍生的任意物质,包括但不限于组织、血液、尿液、皮肤、骨髓、肌肉、毛发、分泌物和内脏器官等。
相关数据[11]:指生物样本的附属信息,包括但不限于研究数据、表型数据、临床数据、流行病学数据和生物样本处理过程得到的数据等。
申办者:指负责临床试验的发起、管理和提供临床试验经费的个人、组织或者机构。
泛知情同意:又称广泛性知情同意,是知情同意的特殊形式,其特点表现为:以该类研究对受试者的低风险性为基础,研究者尽可能对医疗数据和生物样本应用于将来研究履行“告知责任”;提升医疗数据和生物样本应用于将来研究中的“效用”。适用范围包括:(1)未来的研究采集和储存人体的生物材料及相关数据,但特定用途尚不明确。(2)为未来的研究采集和储存可识别身份的医疗数据,但特定用途尚不明确。(3)收集、储存研究剩余的人体生物材料,用于特定用途尚不明确的未来研究。(4)收集、储存临床诊疗过程中剩余的人体生物材料及相关数据,用于特定用途尚不明确的未来研究。(5)采集、储存临床诊疗过程中产生的医疗数据,包括电子病历、影像学资料和各类临床检验检查数据,用于特定用途尚不明确的未来研究。
3 讨论
3.1 《临床试验生物样本伦理管理指南》编制的必要性
生物样本被美国《时代周刊》列为“改变世界的十大新观念”[16]。随着生物医药产业快速发展与自主创新药物研发开展,制药公司、试验机构甚至第三方公司都开始重视收集保存生物样本,各种肿瘤组织库、干细胞库、基因库、胚胎库、病原微生物库如雨后春笋般出现。随着生物样本库的发展,生物样本相关伦理问题逐渐引起关注,引发学界与公众深刻反思。《永生的海拉》[17]是关于美国黑人妇女Henrietta Lacks(海拉)的生命故事。海拉因宫颈癌而死,她的肿瘤细胞却获得了永生,基于海拉细胞的研究获得五项诺贝尔奖,包括“发现HPV”“发现及开发绿色荧光蛋白质”等创造了医学奇迹和巨大商业利益,但是其中涉及的医学伦理与个体尊严等问题引人深思。生物技术领域发展迅猛为重大疑难疾病诊疗带来突破,但是生物技术是把双刃剑,在给人类带来福祉的同时,存在巨大伦理挑战与风险,基因编辑婴儿、人猴嵌合体、人类大脑类器官移植等不断挑战科研伦理底线,生物样本国际合作不当更是给国家与种族安全带来潜在重大威胁。
近年来,药物和医疗器械以及体外诊断试剂临床试验涉及生物样本的采集、保藏、使用和合作研究越来越普遍,其中涉及的伦理问题值得引起关注,包括:样本采集量大、数量多、超方案采集数据信息、国际合作理由不充分、样本储存使用规定不明、剩余样本处理未明确等。虽然有国务院第717 号令与国际国内相关伦理指南[18-19],但是缺乏专门的指南具体指导临床试验中的生物样本采集、保藏、利用、对外提供、销毁等不同环节的伦理要求,特别是明确申办方、第三方检测机构、样本保藏机构等各方在其中的伦理责任。因此,制定专门的《临床试验生物样本伦理管理指南》迫在眉睫。
3.2 《临床试验生物样本伦理管理指南》编制的科学性
传统的德尔菲法为4 轮,改良后的德尔菲法通常为2轮~4 轮,本研究采用改良的德尔菲法[20],经过两轮反复征询汇总专家意见和建议,充分体现了专家的工作经验和知识水平。本课题工作组根据指定的专家标准邀请国内外知名专家38 名,专家的学历、职称、工作年限结构合理,具有良好代表性。专家的积极系数用回收率表示,积极系数一般不低于50%,超过70%则代表积极度非常好[21],本研究2 轮德尔菲法专家咨询问卷回收率均为92. 10%,表明专家积极程度非常高。同时对指南条目提出具体文字性修改意见的专家参与率分别为92. 10% 和73.68%,表明了专家的参与度高。第二轮专家咨询的结果,所有指标的重要性评分均大于4 分,重要性评分变异系数不超过0.2,协调系数为 0.375,P<0.001,说明咨询专家意见的协调程度好,具有良好的科学性与可靠性。通过两轮德尔菲法专家组调研问卷,召开课题专家共识会议,撰写指南初稿,经举办权威专家内审会,最终形成指南终稿。
3.3 《临床试验生物样本伦理管理指南》的创新点
(1)各方共治责任:做好生物样本提供者权益保护,必须依靠临床试验机构、研究者、伦理委员会、申办方、检测机构和样本保藏机构等多方协同,各方均有责任保护生物样本提供者的权益和安全。共治责任也是符合中共中央办公厅、国务院办公厅印发《关于加强科技伦理治理的意见》精神的与时俱进做法。(2)机构管理体系:临床试验机构是生物样本伦理管理的责任主体,由机构领导、相关行政部门、机构办公室、伦理委员会、研究者等组成。管理体系一方面保障生物样本伦理管理的落实到位,也为实施“泛知情同意”应匹配的机构管理系统奠定基础[22]。(3)伦理审查模式:国内尚没有如何开展生物样本伦理审查的具体操作模式,本指南首次提出临床试验涉及生物样本的伦理审查包括两个部分,一是体系审查,主要审查机构对生物样本的管理体系是否满足伦理要求,审查内容包括:管理制度、设施条件、人员资质、隐私保密、泛知情同意、生物安全等;二是项目审查,主要审查具体临床试验项目中涉及生物样本提供者的保护,审查内容包括:样本提供者人群、获益与风险、样本使用、个人信息保密、数据保护、结果披露、退出研究、联系方式等。体系审查侧重管理,只要符合伦理要求,无需每个项目都进行体系的重复审查;项目审查包含在试验方案和知情同意审查内容中,项目审查还要有跟踪审查。
4 结语
针对生物样本库的关键伦理问题与样本生物技术可能造成的重大伦理风险,立足前期研究基础,借鉴发达国家经验,结合我国临床试验生物样本库样本科学的具体实际,通过德尔菲法进行权威专家论证,形成了国内首个《临床试验生物样本伦理管理指南》,希望能对临床试验生物样本的伦理管理以及利用样本开展生物技术的伦理审查实践提供良好的指导作用。
[致谢(按姓名拼音排序):鲍军、白桦、曹彩、曹国英、陈浪、陈晓云、陈勇川、蔡燕宁、丛亚丽、洪明晃、贺晴、姜柏生、季国忠、吉萍、江一峰、贾艳艳、康辉、阚苏立、孔小红、刘晋、李海燕、李晓玲、陆麒、沙莉莉、孙长安、沈一峰、田蕾、吴翠云、万莉、王洁、王嘉楠、王慧萍、王坚、王美霞、汪金海、伍蓉、许重远、许锋、熊宁宁、奚益群、杨国斌、杨亚军、邹冲、赵俊、张馥敏、张妞、张菁、张涛、周人、翟晓梅、朱燕萍]
猜你喜欢
基层中医药(2020年5期)2020-09-11
法制博览(2020年2期)2020-04-29
网络与信息安全学报(2019年6期)2019-12-13
法制与社会(2017年9期)2017-04-18
电视指南(2016年12期)2017-02-05
Coco薇(2015年12期)2015-12-10
专用汽车(2015年12期)2015-03-01
中国合理用药探索(2012年2期)2012-03-20
中国合理用药探索(2011年9期)2011-03-20
中国合理用药探索(2011年7期)2011-03-20
