
高效液相色谱法测定西咪替丁片有关物质
2022-02-01 10:20傅小红姜伟万婷张思甜
化学分析计量 2022年12期
傅小红,姜伟,万婷,张思甜
(南昌市检验检测中心,南昌 330000)
西咪替丁,也称甲氰咪胍,是人类发现的第一种组胺H2受体拮抗剂,能显著抑制基础和夜间胃酸分泌,也能抑制由食物、组胺、分肽胃泌素和胰岛素等刺激引起的胃酸分泌,并对因化学刺激引起的腐蚀性胃炎、应激性胃溃疡和上消化道出血等的胃病治疗有明显疗效[1-3]。近年来的研究发现,西咪替丁在治疗慢性乙型肝炎、心律失常、预防输血反应、辅助治疗支气管哮喘、上呼吸道感染、雄性激素增多以及癌症等疾病也有一定疗效[4-7]。但长期用药或用药剂量过大时可出现腹泻、眩晕或头痛、肌痉挛或肌痛、皮疹、脱发、性欲减退、泌乳现象等不良反应[8-11]。
西咪替丁的制剂类型涉及片剂、胶囊剂、注射剂、口服液制剂以及口服混悬剂。西咪替丁片于1976年由中美史克制药有限公司开发,并于1977年6 月获FDA 批准上市。目前,国内最常见的制剂剂型为片剂,生产企业有319家,涉及批准文号328个。
有关物质是药品质量研究中关键性的项目之一。原料药的有关杂质在生产制剂过程中全部或部分转移到制剂中,同时在制剂生产、储存、运输过程中,主成分自身发生降解或与辅料相互作用,进一步增加了制剂中杂质的种类与总量。基于安全性和生产实际,允许含限定量无害或低毒的杂质,但对毒性较高的杂质则应严格控制。
现行《中国药典》2020 版[12]及其它西咪替丁片质量标准均未设置有关物质检查项。笔者参照中国药典、美国药典[13]、英国药典[14]和欧洲药典[15],建立了西咪替丁片有关物质准确、可靠的高效液相色谱分析方法,并同时测定6种西咪替丁杂质,通过对实际样品的测定分析,阐明了西咪替丁片有关物质的来源,为西咪替丁片安全性和有效性提供科学依据。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱仪:UltiMate 3000型,美国赛默飞世尔科技公司。
电子天平:(1)BT25S 型,感量0.01 mg,德国赛多利斯科学仪器有限公司;(2)ME204E 型,感量0.1 mg,瑞士梅特勒-托利多仪器有限公司。
pH计:PB-10,德国赛多利斯科学仪器有限公司。
紫外分光光度计:UV-2550 型,日本岛津有限公司。
数控超声波清洗器:KQ-500DB型,昆山市超声仪器有限公司。
西咪替丁对照品:100158-201406,纯度为99.8%(质量分数),中国食品药品检定研究院。
西咪替丁杂质B、西咪替丁杂质C、西咪替丁杂质D、西咪替丁杂质E、西咪替丁杂质G、西咪替丁杂质H(6 种杂质具体分子结构参考英国药典[14])对照品:纯度均大于95%(质量分数),上海鸿永生物科技有限公司。
甲醇:色谱纯,美国赛默飞世尔科技公司。
己烷磺酸钠:分析纯,上海展云化工有限公司。三乙胺:分析纯,福晨(天津)化学试剂有限公司。磷酸:分析纯,西陇化工股份有限公司。
实验室用水均为超纯水。
淀粉、预胶化淀粉、玉米淀粉、蔗糖、糊精、微晶纤维素、羟甲基纤维素、羟丙甲纤维素、羧甲淀粉钠、十二烷基硫酸钠、硬脂酸镁、滑石粉、聚山梨酯80、聚维酮K30、二氧化硅、乙醇和亮蓝17种辅料:浙江前进药业有限公司、安徽金太阳生化药业有限公司。
西咪替丁片:市售。
1.2 色谱条件
色谱柱:Welch XtimateTMC18色谱柱(250 mm×4.6 mm,5 μm,月旭科技股份有限公司);流动相:A相为甲醇-水溶液(0.4 mL 三乙胺、0.86 g 己烷磺酸钠和水780 mL 混合后用磷酸调pH 2.8)(体积比为24∶78),B 相为甲醇;梯度洗脱程序:0~55 min 时A相体积分数为100%,55~60 min时A相体积分数由100%变为75%,60~75 min时A相体积分数保持为75%,75~75.1 min 时A 相体积分数由100%变为75%,75.1~85 min 时A 相积分数保持为100%;流量:1.0 mL/min;检测温度:30 ℃;检测波长:220 nm;进样体积:20 μL。
1.3 溶液的制备
西咪替丁与6 种杂质对照品储备液:分别精密称取6种西咪替丁杂质和西咪替丁对照品约25 mg,置于7只25 mL容量瓶中,分别加入甲醇,超声溶解并定容。各对照品的质量浓度均为1.0 mg/mL。
混合对照品溶液:分别取7 种对照品储备液1 mL,置于10 mL容量瓶中,加入流动相A,定容至标线,摇匀,其中各组分质量浓度均为0.1 mg/mL。
系列混合标准工作溶液:分别量取适量的混合对照品溶液,用流动相A分别稀释,配制成质量浓度为5、10、20、50、80、100、150、200 μg/mL系列混合标准工作溶液。
西咪替丁片样品溶液:西咪替丁的质量浓度为0.4 mg/mL。取西咪替丁片样品20片,研细,精密称取细粉适量(约相当于西咪替丁0.2 g),置50 mL 容量瓶中,加甲醇10 mL超声使其溶解,再加流动相A定容至标线,摇匀,过滤,精密量取续滤液5 mL,置于50 mL容量瓶中,用流动相A稀释至标线,摇匀。
辅料空白溶液:取17 种辅料,均称取12.5 mg,置于同一只25 mL容量瓶中,加入10 mL甲醇,超声使溶解,再用流动相A定容至标线,摇匀,作为辅料空白储备液。量取1 mL 辅料空白储备液,置于5 mL容量瓶中,加入流动相A稀释至标线。
1.4 实验步骤
按1.2 色谱条件测定样品溶液及加标回收样品溶液,利用色谱峰面积标准曲线法计算出西咪替丁及6种杂质的含量。
2 结果与讨论
2.1 检测波长的选择
分别取1.3 中7 种对照品储备溶液稀释至合适浓度,在190~400 nm波长进行扫描,测得西咪替丁杂质B、西咪替丁杂质C、西咪替丁杂质D、西咪替丁杂质E、西咪替丁杂质G、西咪替丁杂质H以及西咪替丁的最大吸收波长分别为219、196、195、217、220、220、220 nm,综合考虑,最终选择检测波长为220 nm。
2.2 色谱条件的优化
参考国内外药典标准[12-15],笔者最初选择欧洲药典所提供的色谱条件,A 相为甲醇-水溶液(0.4 mL 三乙胺、0.86 g 己烷磺酸钠和水780 mL 混合后用磷酸调pH 2.8)(体积比为24∶76),B 相为甲醇;按梯度洗脱,洗脱程序:0~60 min 时A 相体积分数为100%,60~65 min 时A 相体积分数由100%过渡到90%,65~120 min时A相积分数保持为90%。但按上述条件进行实验耗时过长,因此在此基础上对实验条件进行优化改良,最终确认了1.2 中的色谱条件。结果表明,在不影响有关物质分析的前提下,按1.2 实验条件所得的色谱峰峰形更好,分离度更高,且耗时更短。取混合对照品溶液,按1.2的色谱条件进行分析,记录色谱图,西咪替丁与各杂质的分离度均大于1.5,各色谱峰峰形对称且理论塔板数均大于5 000。图1 为7 种化合物混合剂对照品溶液色谱图。
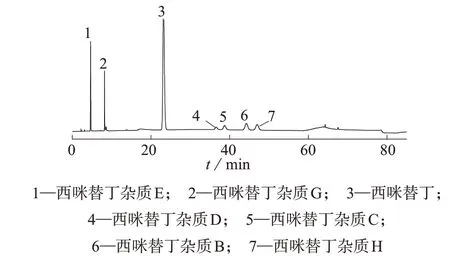
图1 7种化合物混合对照品溶液色谱图
2.3 专属性实验
取1.3 中辅料空白溶液按1.2 色谱条件进样测定,所得的色谱图见图2。结果显示,辅料空白溶液出峰时间集中4分钟之前,且峰面积很小,保留时间为62~65 min的三个色谱峰为亮蓝的特征色谱峰,与混合对照品溶液色谱图对比发现,辅料组成对样品溶液有关物质的测定不产生干扰。
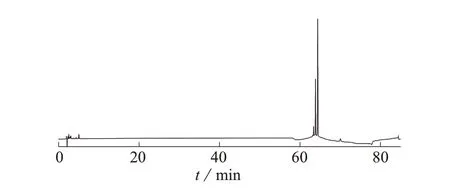
图2 辅料空白溶液色谱图
2.4 线性方程与检出限
取1.3中的混合标准工作溶液按照1.2色谱条件进行测定,以各成分的质量浓度(x)为横坐标,以色谱峰的峰面积(y)为纵坐标,绘制标准工作曲线,计算线性方程和相关系数。将1.3 中的混合对照品溶液用流动相A 逐级稀释,按照1.2 色谱条件进行测定,以3倍信噪比对应的质量浓度作为检出限,所得结果见表1。

表1 7种化合物质量浓度线性范围、线性方程、相关系数及检出限
2.5 稳定性试验
取混合对照品溶液分别于制备后0、4、8、12、16、20、24 h按1.2色谱条件进行测定,记录色谱峰面积。各化合物的相对偏标准差见表2,由表2可知,7种化合物峰面积的相对标准偏差为0.42%~0.96%,表明该方法稳定性良好。
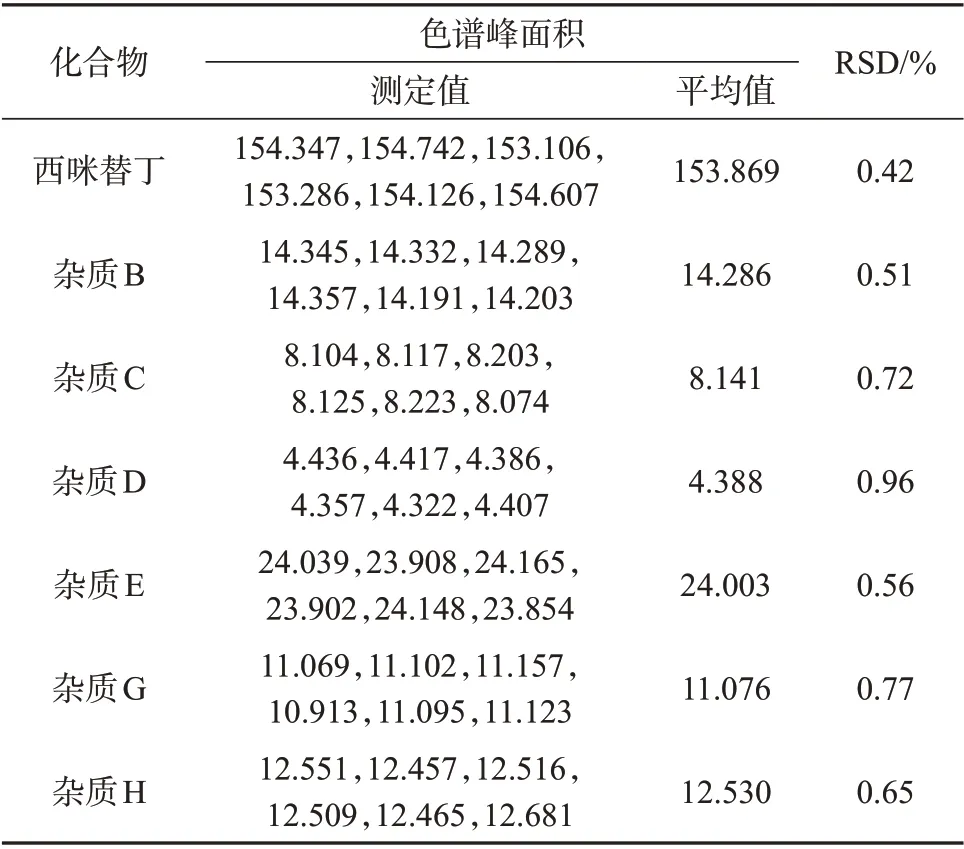
表2 稳定性试验结果
2.6 加标回收与精密度试验
分别取1.3中辅料空白储备液1 mL,置于9只5 mL容量瓶中,其中3只分别加入0.2 mL混合对照品溶液,3 只分别加入0.4 mL 混合对照品溶液,3 只分别加入0.6 mL混合对照品溶液,均用流动相A定容至标线,按1.2色谱条件分别进行测定,加标样品色谱图见图3,测定结果见表3。由表3可知,7种化合物平均回收率为97.3%~102.2%,测定结果的相对标准偏差为0.92%~3.32%,表明该方法重复性好,准确度高。
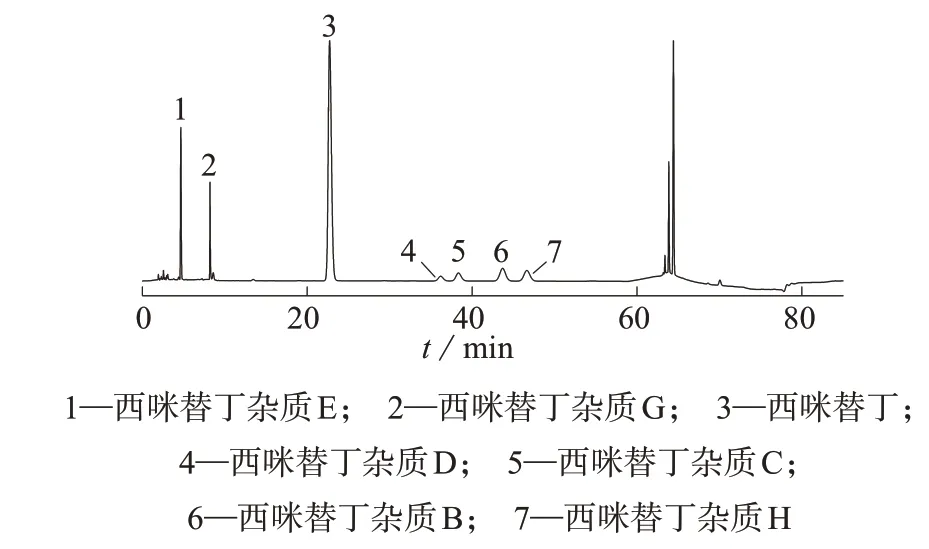
图3 加标样品色谱图
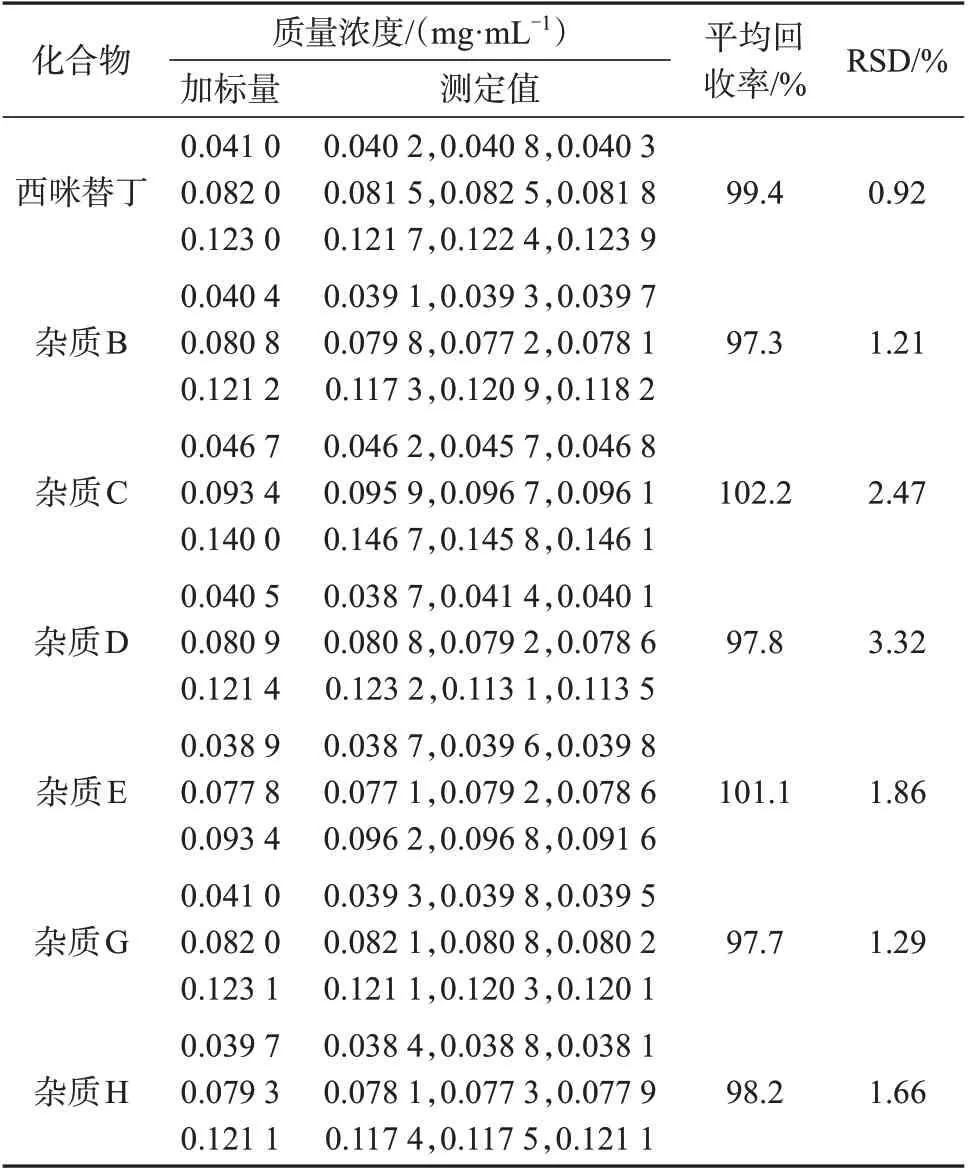
表3 加标回收与精密度试验结果
3 结语
建立了西咪替丁片有关物质的高效液相色谱测定方法,研究表明,所建立的方法稳定性好、准确度高、专属性强。该方法为科学评价西咪替丁片有关物质和品种配方工艺优化提供可靠的实施途径,具有重要的研究和实际应用价值。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
中国应急管理科学(2021年4期)2021-04-13
纺织服装流行趋势展望(2020年1期)2020-02-01
中学生数理化·高一版(2020年9期)2020-01-02
中学化学(2019年4期)2019-08-06
考试周刊(2018年68期)2018-09-17
纺织服装流行趋势展望(2016年6期)2016-05-04
纺织服装流行趋势展望(2016年4期)2016-05-04
纺织服装流行趋势展望(2016年1期)2016-05-04
家庭医学(2016年3期)2016-04-05
