
首诊于眼科的IgG4相关疾病初步研究△
2022-01-19 12:14付俊霞王永平许光璨宋宏鲁孙明明魏世辉周欢粉
中国眼耳鼻喉科杂志 2022年1期
付俊霞 王永平 许光璨 宋宏鲁 孙明明 魏世辉 周欢粉
(1.解放军总医院第一医学中心眼科 北京 100853;2.解放军总医院第三医学中心眼科医学部 北京 100853)
IgG4相关疾病(IgG4 related disease,IgG4-RD)是一种近年来认识的自身免疫性疾病,以血清IgG4水平显著升高和组织或器官肿块样病灶为最常见的临床表现,其主要的组织病理表现为IgG4+浆细胞为主的淋巴细胞、浆细胞浸润,并伴有席纹状纤维化、闭塞性静脉炎和嗜酸性粒细胞浸润[1-3]。该病几乎可以影响任何器官系统,经典的受累部位是唾液腺、胰腺、泪腺、肝胆道、眼眶及眶内组织、淋巴结和腹膜后等[1]。不同脏器受累患者临床特点差异较大,IgG4相关眼病(IgG4-related ocular disease,IgG4-ROD)是最近描述的一种疾病,眼睑、泪腺、眼外肌、眼眶软组织、巩膜、视神经、眶神经及眶骨均可受影响[4]。目前关于IgG4-ROD的流行病学数据很少,来自日本的数据估计该国的发病率为每10万人中有0.28~1.08人次,每年新增336~1 300例患者[5]。关于IgG4-ROD的文章系统性观察研究相对较少,为此我们对一组诊断为IgG4-ROD患者的临床资料进行总结分析,现将其临床表现、实验室检查、影像学检查、治疗及预后特征报告如下。
1 资料与方法
1.1 资料 回顾性病例分析。本研究经解放军总医院伦理委员会审核批准(批准号:S2017-093-01),严格遵循《赫尔辛基宣言》,取得所有患者的知情同意。
选取2011年1月~2021年4月解放军总医院第一医学中心神经眼科诊断为IgG4-ROD的9例患者。IgG4-ROD诊断标准参考2015年日本学者[6]。(1)影像学检查:泪腺增大、三叉神经或眼外肌的增粗,以及各种眼眶组织出现肿大或增生性病变。(2)组织病理学检查:①明显的淋巴细胞和浆细胞浸润,伴纤维化;②可观察到生发中心;③IgG4+浆细胞/IgG+浆细胞>40%,或IgG4+浆细胞>50个/HPF(高倍镜视野);(3)血液检查:血清IgG4 升高(≥135 mg/dL)。满足(1)+(2)+(3)可确诊;满足(1)+(2)为拟诊;满足(1)+(3)为疑诊。
1.2 方法 总结9例IgG4-ROD患者的临床表现、实验室检查、眼眶磁共振成像(magnetic resonance imaging,MRI)、病理学检查和治疗情况。
1.3 统计学处理 应用SPSS26.0软件对数据进行分析,对样本量大于3的定量资料进行正态性检验,服从正态分布者采用均数±标准差表示,不服从正态分布者采取中位数和四分位数表达。
2 结果
9例患者,男性6例、女性3例,就诊年龄为31~60岁,平均年龄为(48.67±10.36)岁,病程为20 d~12年,2例患者累及单眼,余均为双眼受累。IgG4-ROD确诊6例,拟诊2例,疑诊1例。就诊时眼睑肿胀6例,眼球突出4例,上睑下垂1例,复视1例,视力下降5例,有1例患者(病例4)同时伴有听力下降、牙痛和面颊肿胀(表1)。

表1 9例患者临床表现
2例患者伴有哮喘(病例5和6),2例伴有高尿酸血症(病例2和9)。2例累及颌下腺和腮腺(病例1和3),1例累及颌下腺、胰腺和颈部淋巴结(病例2)。病例8颌下腺肿大,病例4累及腮腺。
8例患者进行了红细胞沉降率(erythrocyte sedimentation rate,ESR)检查,6例升高,高者可达94 mm/h(64.23~76.50 mm/h)[正常值:0~20 mm/h(女);0~15 mm/h(男)]。9例患者中仅1例患者C-反应蛋白(C-reactive protein,CRP)升高(病例9:9.97 mg/dL,正常值:0~0.8 mg/dL)。8例患者IgG4均有不同程度的升高,最高可达8 430 mg/dL(中位数:1 610 mg/dL;P25~P75:1 560~4 690 mg/dL;正常值:3~201 mg/dL)。8例患者中,4例IgG1增高,高者可达2 590 mg/dL(中位数:1 150 mg/dL;P25~P75:844.50~1 630 mg/dL;正常值:405~1 011 mg/dL);9例患者中,1例IgG2增高(病例2:883 mg/dL;正常值:169~786 mg/dL),2例IgG3增高(病例1:186 mg/dL;病例9:345 mg/dL;正常值:11~85 mg/dL)。
所有患者IgA浓度均正常,8例IgE浓度升高,最高者可达2 360 IU/mL(中位数:905 IU/mL;P25~P75:231.50~1 121.00 IU/mL;正常值:0~100 IU/mL),7例IgG浓度升高,可达7 460 mg/dL(中位数:3 140;P25~P75:1 760~3 635 mg/dL;正常值:700~1 600 mg/dL),2例IgM浓度降低(病例1:37.2mg/dL;病例6:33.4 mg/dL;正常值:40~230 mg/dL)。3例患者血清补体C3浓度降低(病例1、3、6,分别为67.9、82.8、70.8 mg/dL;正常值:90~180 mg/dL);2例C4浓度降低(病例1和3,分别为7.63、9.45 mg/dL);1例C4浓度升高(病例7:45.2 mg/dL;正常值:10~40 mg/dL)。
3例患者住院期间行腰椎穿刺及脑脊液(cerebrospinal fluid,CSF)检查(病例2、4、7),结果显示:病例2 CSF蛋白和IgG水平升高,分别为423.9、6.73 mg/L;病例4 CSF白细胞、蛋白、IgG、IgM升高,依次为 200×106个/L、461.1 mg/L(正常值:150~400 mg/L)、10.9 mg/L(正常值:0~3.4 mg/dL)、1.81 mg/dL(正常值:0~0.13 mg/dL)。
9例患者行抗核抗体5项及自身抗体谱11项检查,6例患者行类风湿3项检查,结果均为阴性。
9例患者中,8例行眼眶MRI平扫+增强,多表现为双侧泪腺弥漫性增大,伴或不伴眼外肌增粗,等T1信号,稍长T2信号,增强后明显强化(图1)。3例患者眼球突出,7例患者眼外肌受累,5例泪腺受累,3例视神经受累,本研究中尚未发现三叉神经分支受累者(表2)。
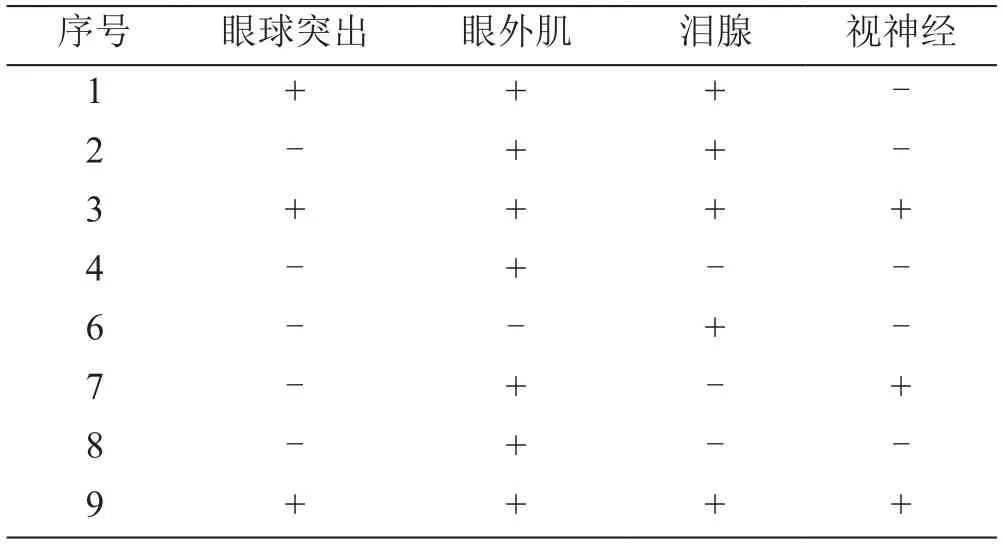
表2 8例患者眼眶MRI累及部位

图1 眼眶MRI 患者1右眼眼球突出,双眼内直肌、右眼上直肌明显增粗(白箭),多组鼻旁窦炎并左侧上颌窦黏膜下囊肿。A.水平位T1WI双眼内直肌呈等T1信号;B. 水平位T2WI双眼内直肌、右眼上直肌稍长T2信号;C. 水平位T1WI增强双内直肌明显强化;D. 冠状位T2WI 双眼内直肌、右眼上直肌稍长T2信号;E. 冠状位T1WI增强后双眼内直肌、右眼上直肌明显强化。
8例患者有组织病理活检结果(图2),均有不同程度纤维组织增生及伴多量淋巴细胞、浆细胞浸润,其中5例IgG4/IgG>40%。由于不同病理医师习惯性表述差异,5例IgG4+细胞阳性,其中1例>50个/HPF,1例>30个/HPF,余未给出具体细胞数(表3)。

图2 光镜下患者9泪腺组织 A.HE染色(×10):增生的淋巴组织伴较多淋巴滤泡形成,间质纤维组织增生,并分隔淋巴组织,伴多量淋巴细胞、浆细胞浸润。B.免疫组织化学染色(×10):IgG4(+),局部>50个/HPF。

表3 8例患者病理学结果
9例患者除1例拒绝激素冲击治疗外,余均给予250 mg~1 g激素冲击联合营养神经改善循环治疗,同时结合患者实际情况选择他克莫司(病例7和9)或者利妥昔单抗(RTX)(病例5和6)来抑制复发。患者眼睑肿胀,眼球突出(病例7入院时眼球突出度35/25 mm,出院时24/18 mm),眼球运动情况均有不同程度的好转。在5例有视力下降主诉的患者中,4例患者患眼或对侧眼视力均有不同程度的好转(病例3、4、8和9),1例患者视力维持稳定(病例2),详见表4。
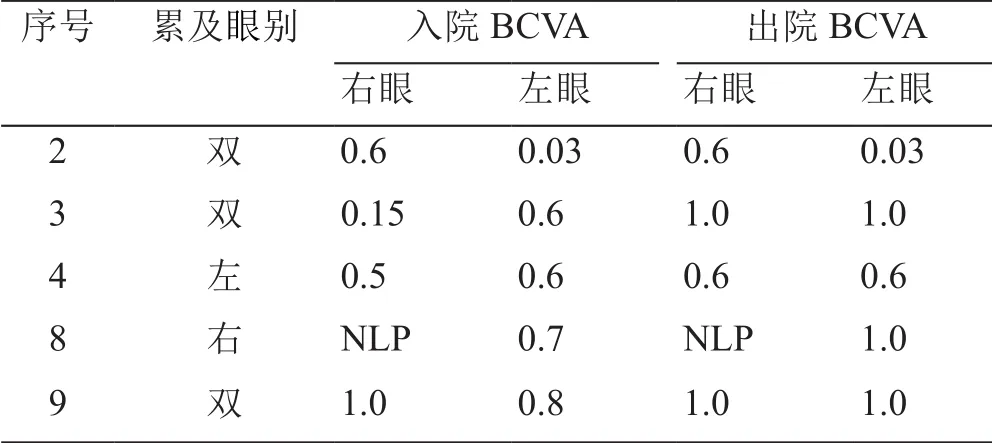
表4 5例患者出入院视力情况
3 讨论
2001年,Hamano等[7]报告了自身免疫性胰腺炎患者血清IgG4水平异常升高,随后开始了IgG4与全身疾病相关研究。除胰腺炎外,泪腺、唾液腺、肝胆道和腹膜后出现的器官肿大或肥厚患者中均可出现血清IgG4水平升高。随后相关研究[8]发现,该组疾病具有IgG4阳性浆细胞浸润,局部纤维化,伴滤泡形成的病理特征。眼部泪腺和唾液腺对称肿大的患者,过去常常被诊断为Mikulicz病[9]。直到2012年日本学者提出IgG4-RD的诊断标准,二者才被认为是不同的疾病,并对IgG4相关肾病和胰腺病提出了相应的诊断标准[2],而IgG4-ROD直到2014年才被提出[1]。
IgG4-ROD目前被认为与遗传、感染、免疫相关,IgG4抗体可通过交换重链和附加轻链来交换Fab臂,可形成双特异性抗体,也可作为单价分子发挥作用[10],其产生需要Th2细胞辅助。Zen等[11]研究发现,IgG4相关性硬化胰腺炎和胆管炎患者外周血和受累器官中CD4+CD25+Foxp3+调节T细胞显著增多,认为其可能的机制是调节性T细胞和Th2免疫平衡打破,进而产生一系列炎症因子(IL-10和TGF-β等),导致组织内大量IgG4+浆细胞浸润和组织纤维化。
3.1 临床表现 该病的主要临床特征是双侧泪腺对称性无痛性增大伴多条眼外肌肿胀、眶下神经增粗和压迫性视神经病变。临床上患者多以眼部肿胀、眼球突出伴或不伴视力下降就诊,中老年人常见,在眼部男女患病比例相当,无明显的性别倾向。本研究中,男性受累居多(6例,66%),就诊平均年龄为(48.67±10.36)岁,双眼受累多见(77.8%),以眼睑肿胀、眼球突出、视力下降就诊多见,累及眼外肌时可出现复视症状,还可伴有耳面部症状。
IgG4-ROD患者可伴有全身其他免疫性疾病,可有过敏史。本研究中2例患者伴有哮喘,2例伴有高尿酸血症,部分患者累及颌下腺、腮腺、胰腺和颈部淋巴结。
IgG4-ROD多数患者血清IgG4水平升高(>1 350 mg/L),但是血清IgG4高水平并非IgG4-RD特异性指标,其他疾病系统性红斑狼疮、恶性肿瘤亦可出现IgG4水平升高[12],大约30%的IgG4-RD患者血清IgG4水平正常[13]。由于IgG4-RD临床表现为多样,目前任何单一指标均无法对患者进行准确诊断和分类,血清IgG4水平结合组织病理学及临床特征对IgG4-RD有较好的诊断价值[14]。本研究中8例患者IgG4均有不同程度的升高,大部分患者血清IgE水平升高(8例),部分患者合并IgG其他亚型及补体水平异常,2例患者存在不同程度的脑脊液IgG和IgM异常,提示在本病发病过程中存在免疫紊乱。有文献[16]报道,该病可合并抗核抗体、抗中性粒细胞抗体、类风湿因子等风湿免疫抗体异常。急性期ESR可升高,本研究中6例ESR升高,最高者可达94 mm/h,提示病情较重。
IgG4-ROD典型的影像学表现为双侧泪腺弥漫性对称性肿大、眶内边界清楚密度均匀的软组织肿块、双侧眶下神经或眼外肌增粗,MRI T1WI呈等或稍低信号,T2WI多呈等或稍高信号,增强扫描明显均匀强化[15],双泪腺肿大合并眶下神经增粗对于诊断有较高的特异性。本研究中样本量相对较少尚未发现眶下神经增粗的病例,大部分患者MRI表现为泪腺和(或)眼外肌受累,伴或不伴视神经受压。
IgG4-RD典型的病理特征如下。①大量的淋巴浆细胞浸润。②席纹状纤维化:梭形细胞车轮状由中心向四周放射状排列。③闭塞性静脉炎:由大量的淋巴细胞和浆细胞浸润静脉致管腔部分或完全闭塞。该特征在IgG4-ROD相对少见,目前被认为是一种器官特异性的表现,而不是某种特定疾病的病理特征[3,16]。不同受累组织中IgG4+浆细胞/HPF迄今尚未统一,多数建议IgG4+浆细胞/HPF>30~40个,但在腹膜后纤维化或一些肾脏病变,IgG4+浆细胞/HPF>10个即可认为阳性,IgG4+浆细胞/IgG+浆细胞>40%亦是诊断IgG4-RD的重要依据[1]。由于IgGROD可处于不同的时期,故病理切片检查结果可不尽相同。
3.2 鉴别诊断 本病尚需和眼眶的其他疾病,如眼眶黏膜相关淋巴组织结外边缘区B细胞淋巴瘤(MALT淋巴瘤)、良性淋巴上皮病变(Mikuliczs病)、特发性眼眶炎性假瘤、甲状腺相关性眼病(Graves病)进行鉴别。
(1) MALT淋巴瘤是一类恶性程度低的B细胞非霍奇金淋巴瘤,该类患者可伴有IgG4+浆细胞数量增多和IgG4浓度升高[17]。二者鉴别存在一定困难,此类患者病理除浆细胞浸润外,还伴有免疫球蛋白轻链限制或免疫球蛋白基因重排,分子生物学检查有助于鉴别两者。
(2) Mikuliczs病表现为泪腺慢性硬化并伴有腮腺、颌下腺肿大,既往因多数患者表现出唾液及泪液分泌减少,口干、眼干的症状而曾经被认为是干燥综合征(SS)的亚型。2008年日本干燥综合征协会提出IgG4+的Mikuliczs病的诊断标准[18-19],近年来随着对IgG4-ROD的研究更加深入,Mikuliczs病被确定为IgG4-ROD的一种亚型,可合并自身免疫性血小板减少[20]。
(3) 特发性眼眶炎性假瘤又称眼眶非感染性非特异性炎性反应,病理表现为多种炎症细胞浸润和不同程度纤维化的非特异性炎性反应。特发性眼眶炎性假瘤患者多伴有明显的疼痛,IgG4-ROD多慢性起病,双侧泪腺受累多见,很少出现眼部疼痛,多伴有全身其他系统疾病。
(4) Graves病可合并甲状腺功能亢进、正常或低下,常以复视和眼球运动异常为主诉就诊,肌肉受累频度依次为下直肌、内直肌、上直肌、外直肌,且CT表现为肌腹肥厚,肌肉止点多属正常,结合全身检查及影像学检查有助于鉴别。
3.3 治疗 针对IgG4-RD目前没有明确统一的治疗标准。2019年Detiger等[21]发表的系统综述中纳入35篇95例IgG4-ROD患者,95患者79例首选糖皮质激素治疗,89%患者对糖皮质激素治疗有反应。在接受糖皮质激素治疗的83例患者中,12例(14%)在糖皮质激素减量时复发,30例(36%)在停用糖皮质激素后复发。给予利妥昔单抗(rituximab,RTX,59%)、甲氨蝶呤(27%)、硫唑嘌呤(15%)、吗替麦考酚酯(11%)和(或)其他生物制剂。结果表明,传统抗风湿药(disease-modifying antirheumatic drugs,DMARD)的成功率为36%~75%,而RTX成功率为93%,提示RTX是治疗IgG4-ROD最有效的生物制剂。传统DMARD疗效有限,并建议IgG4-ROD在难治性、危及重要脏器或生命时,尽早开始RTX治疗。2021年《IgG4-RD诊治中国专家共识》[1]中提到:①有症状且病情活动的IgG4-RD患者应接受治疗,无症状但重要脏器受累并进展的患者亦需及时治疗;②糖皮质激素是治疗IgG4-RD的一线药物,小剂量激素维持治疗可降低复发率,维持治疗时间推荐1~3年;③免疫抑制剂与糖皮质激素联合使用较单用糖皮质激素更有效控制疾病,减少IgG4-RD患者的复发;④难治性或复发性IgG4-RD可选用生物制剂;⑤当IgG4-RD患者特殊部位受累,可能引起压迫等导致器官功能障碍等紧急情况,如药物治疗不能迅速解除时,需采取快速、有效的外科手术或介入治疗进行干预,尽快缓解症状,避免病情进一步恶化,为后续药物治疗创造条件。本研究中除1例拒绝激素冲击治疗外,急性期均给予大剂量甲泼尼龙冲击治疗,患者眼睑肿胀、眼球突出、复视及视力情况均有不同程度缓解。因此,笔者建议急性期给予大剂量激素冲击治疗,小剂量激素联合免疫抑制剂治疗预防其复发,压迫或累及重要脏器时也可行手术或介入治疗。
本研究为单中心描述性研究,缺少随访,病例数也有限。鉴于此,IgG-ROD仍需眼科医师给予更多关注并进行大样本统计分析,总结其临床特征,使患者得到更好的救治。
猜你喜欢
畜牧兽医学报(2022年8期)2022-08-26
中国临床医学(2022年3期)2022-07-08
中国典型病例大全(2022年9期)2022-04-19
现代检验医学杂志(2022年2期)2022-04-19
家庭医药(2021年7期)2021-07-23
中国现代医生(2021年8期)2021-04-30
湖南中医药大学学报(2019年4期)2019-09-10
中国医药导报(2019年5期)2019-04-28
妇女之友(2018年1期)2018-03-02
养生大世界(2016年4期)2016-09-19
