
海藻糖脂肪酸二酯的酶法合成及性质
2022-01-06 05:01汪淑珍
食品科学 2021年24期
汪淑珍
(暨南大学食品科学与工程系,广东 广州 510632)
海藻糖是一类广泛分布于虾、藻类及酵母等生物中的天然二糖,其结构以两分子葡萄糖通过α,α-1,1-糖苷键连接而成[1]。除了作为各类生物的结构及能量物质外[2],海藻糖还具有一些极佳的功能性质。例如,马路凯等[3]在冷冻熟制水产品中添加海藻糖作为保水剂,发挥抗冻保水的作用;Vílchez等[4]对土豆和辣椒接种一组耐干旱微生物菌株,通过该菌株产生的海藻糖可保护植物免受干旱,从而使植物具有抗干旱的能力;Lee等[5]研究发现蛋白类药物中添加海藻糖水凝胶可维持其在运输和长期贮藏过程中的稳定性。同时,海藻糖的亲水性羟基易与各类脂肪酸构成海藻糖酯,提升其乳化[6]、胶凝[7-8]、流变[9]及赋形[10]等理化性能,以及抗菌[11-12]、抗生物膜形成[12]、抗肿瘤[13]、抗氧化[2,14]及抗炎[11]等生物活性,并且海藻糖酯自身无毒无刺激、易降解等特性,扩大了其在食品[9]、药品[15-17]、化妆品[18]等领域的应用范围。
海藻糖酯可通过酯化或酯交换反应进行化学或酶催化合成。其中,化学催化合成因反应过程中酰氯或毒性偶联剂的残留、苛刻的工艺参数及副产物的产生等不利因素逐渐被摒弃[11]。Paul等[19]以2-(1H-苯并三偶氮-1-基)-1,1,3,3-四甲基脲四氟硼酸酯(2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate,TBTU)为偶联剂合成海藻糖单酯和二酯的混合物,不仅分离纯化困难,而且TBTU的使用也容易造成环境危害。而酶催化合成因具有反应条件温和、选择性高和操作简单等优点引发了研究者们极大的兴趣[20]。在酶种类筛选过程中发现,脂肪酶[6]和蛋白酶[21]具有选择特异性及催化效率高等特点,常作为酶促反应催化剂用于海藻糖酯的合成。其中,脂肪酶通常选择性作用于己糖苷的伯羟基合成特异性糖酯[22],且广泛来源于植物、动物和微生物[23]。据文献报道,微生物来源的南极假丝酵母脂肪酶B(Candida antarcticalipase B,CALB)常用于海藻糖酯的合成并获得较高的得率[8,22]。而固定化CALB如固定化脂肪酶Novozym 435[14,24]和Fermase CALB™ 10000[11]等不仅易于合成海藻糖酯,而且还易于从反应混合物中分离[25]。在固定化脂肪酶催化下合成糖酯时,由于糖类和脂肪酸直接酯化脱水缩合生成副产物水,而极少量的水亦能在酶表面形成单层水,干扰酶活性位点的底物结合,因此水起到竞争性抑制剂作用,促进水解这一逆反应,导致得率降低[6,26]。Marathe等[11]通过酯化反应将海藻糖转化为其棕榈酸酯,转化率仅为35%,且产物为单酯和二酯的混合物,还需要后续分离和纯化。
因此,目前常用的海藻糖酯合成方法,是通过海藻糖与脂肪酸乙烯酯发生不可逆的酯交换反应,即以脂肪酸乙烯酯为酰基供体,生成易从反应介质中除去的乙醛(在室温下为气体),有效使反应平衡向酯生成方向移动,具有较高的反应速率和得率[27-28]。据报道,Hibert等[8]使用合成的脂肪酸乙烯酯与海藻糖发生酯交换反应生成海藻糖脂肪酸二酯,得率可达到50%。对于酯交换反应,有机溶剂等非水介质的加入通常有利于反应的进行,然而,极性有机溶剂虽提高了反应底物的溶解度,但易使酶失活,而非极性有机溶剂虽有助于保持酶活力但会使二糖溶解度降低。为了解决这一矛盾,可采用混合溶剂以兼顾糖类反应物的溶解性和酶活力的保持。Chen Jie等[29]在吡啶和叔丁醇(4∶6,V/V)的混合溶剂中,通过CALB催化合成海藻糖亚油酸单酯的产物浓度可达近20 mmol/L。
本研究拟通过在混合溶剂中,探究海藻糖和常见C6~C18脂肪酸乙烯酯在固定化脂肪酶催化下选择性合成相应6,6’-O-海藻糖脂肪酸二酯的最佳条件。通过核磁共振波谱和质谱分析鉴定产物结构,测定系列海藻糖脂肪酸二酯的亲水亲油平衡(hydrophilic lipophilic balance,HLB)值、发泡性能、热稳定性以及细胞毒性,以期为海藻糖脂肪酸二酯的合成提供一种绿色有效的新方法,评价其作为稳定的O/W型食品乳化剂的应用前景,为其综合利用原料和开发具有价值的新型糖酯提供新的研究方向。
1 材料与方法
1.1 材料与试剂
鼠巨噬细胞RAW267.4、人高转移肝癌细胞HCCLM3、人肝癌细胞HepG2、宫颈癌细胞Hela、人肺癌细胞A549、人乳腺癌细胞MCF-7 中国科学院细胞库。
乙酸乙烯酯、己酸乙烯酯、辛酸乙烯酯、癸酸乙烯酯、月桂酸乙烯酯、肉豆蔻酸乙烯酯、棕榈酸乙烯酯、Span 20、油酸、醋酸钯 东京化学工业有限公司;海藻糖、吡啶、叔丁醇、阿霉素 萨恩化学技术(上海)有限公司;Novozym 435固定化脂肪酶 诺维信生物技术(上海)有限公司;氢氧化钾 天津市福晨化学试剂厂;蔗糖酯S-1170 日本三菱化学食品公司:柱层析硅胶、薄层层析板 青岛海洋化工有限公司;石油醚、乙酸乙酯、二氯甲烷、甲醇(均为分析纯) 广州市光华科技股份有限公司;氘代氯仿、氘代甲醇 美国CIL公司。
1.2 仪器与设备
EL104电子天平 梅特勒-托利多(上海)有限公司;MS-H-Pro磁力搅拌器 美国赛洛捷克公司;N-1300型旋转蒸发器 东京理化器械株式会社;AVANCE III型核磁共振(nuclear magnetic resonance,NMR)波谱仪(300、400、500 MHz和600 MHz) 瑞士布鲁克公司;4000 Q-Trap-质谱(mass spectrometry,MS)仪 美国应用生物系统公司;T25高速剪切机 德国IKA公司;TG209F3-ASC热重分析仪 德国耐驰公司;96 孔聚苯乙烯微孔板 美国康宁公司;SynergyTMHT多功能酶标仪 美国博腾仪器有限公司。
1.3 方法
1.3.1 油酸乙烯酯的合成
1.3.1.1 化学合成油酸乙烯酯
在500 mL圆底烧瓶中依次加入36.2 mmol油酸、140.0 g乙酸乙烯酯、0.04 g醋酸钯、0.2 g氢氧化钾。室温、氮气保护下混合搅拌反应30 h(图1)[30]。

图1 油酸乙烯酯的化学合成Fig.1 Chemical synthesis of oleic acid vinyl esters
1.3.1.2 薄层色谱(thin layer chromatography,TLC)法定性分析油酸乙烯酯
TLC法是根据不同化合物极性不同,通过流动相时迁移速率不同,从而将样品中各组分按极性大小展开,通过显色剂显色后测定各组分斑点的迁移率(Rf)定性分析目标产物。
用毛细管取反应结束后的混合物和经硅胶柱层析收集的粗分离液,在薄层层析板上点样,以体积比9∶1的石油醚-乙酸乙酯为展开剂,展开至溶剂前沿取出,待展开剂挥发完后,用碱性高锰酸钾显色,斑点为亮黄色。按式(1)计算各组分斑点的Rf值:

1.3.1.3 硅胶柱层析分离纯化油酸乙烯酯
反应结束后,先在45 ℃下旋转蒸发除去多余的乙酸乙烯酯,再取硅胶,用石油醚湿法装柱,用体积比490∶10的石油醚-乙酸乙酯梯度洗脱,TLC监测,收集目标产物,浓缩得到所需的油酸乙烯酯。按式(2)计算油酸乙烯酯得率:

1.3.1.4 油酸乙烯酯结构鉴定
1H NMR、13C NMR检测条件:氘代氯仿为溶剂,四甲基硅烷为内标,25 ℃下分别采用300 MHz和101 MHz进行数据收集。
1.3.2 海藻糖脂肪酸二酯的合成
1.3.2.1 酶法合成海藻糖脂肪酸二酯
参考Li Xuan等[31]的棉子糖脂肪酸酯合成方法并加以改进。将2.0 g海藻糖溶解于65 mL无水吡啶中,然后添加80 mL预热至60 ℃的无水叔丁醇;加入2.0 g固定化脂肪酶Novozym 435,并在60 ℃下通过磁力搅拌器搅拌悬浮液(250 r/min、30 min);最后按照海藻糖和脂肪酸乙烯酯物质的量比为1∶4加入相应的脂肪酸乙烯酯(23.2 mmol),反应72 h,具体过程如图2所示。

图2 海藻糖脂肪酸二酯的酶促合成Fig.2 Enzymatic synthesis of trehalose fatty acid diesters
1.3.2.2 TLC法定性分析海藻糖脂肪酸二酯
按1.3.1.2节步骤用TLC法进行海藻糖脂肪酸二酯定性分析,展开剂为体积比4∶1的二氯甲烷-甲醇。
1.3.2.3 硅胶柱层析分离纯化海藻糖脂肪酸二酯
反应结束后,先在55 ℃下旋转蒸发除去多余溶剂,再过滤浓缩得到黄色油状粗产物,最后取硅胶,用二氯甲烷湿法装柱,体积比9∶1的二氯甲烷-甲醇溶液梯度洗脱,TLC监测,收集目标产物,浓缩得到所需的海藻糖脂肪酸二酯。按式(3)计算海藻糖脂肪酸二酯得率:

1.3.2.4 海藻糖脂肪酸二酯结构鉴定
1H NMR、13C NMR检测条件:氘代甲醇为溶剂,25 ℃下分别采用600 MHz和151 MHz(海藻糖棕榈酸二酯分别采用500 MHz和126 MHz)进行数据收集。
用单四极杆液相色谱-MS仪用于获取低分辨率MS。MS条件:电喷雾电离(electron spray ionization,ESI)离子源,正离子模式检测,气帘气压力20 psi,去溶剂温度0 ℃,雾化气压力20 psi,加热气压力0 psi,喷雾电压5 500 V,去簇电压50 V,射入电压10 V;质量扫描范围m/z50~1 800;扫描时间1.750 s。
1.3.3 海藻糖脂肪酸二酯的HLB值计算
HLB值由表面活性剂分子中所含的亲水基团和疏水基团的物质的量决定。按式(4)计算海藻糖脂肪酸二酯的HLB值[32]:

1.3.4 海藻糖脂肪酸二酯的起泡性和泡沫稳定性测定
称取一定量的系列海藻糖脂肪酸二酯溶液及对照物S-1170和Span20于100 mL离心管中,25 ℃下用蒸馏水配制质量浓度均为0.2 g/100 mL的溶液,记录溶液起始高度(h0)。用高速剪切机于2 000×g下分散均质2 min,立即测定每组样品的泡沫高度(h2)和液体总高度(h1),分别于静置20、40、60 min和80 min后记录泡沫高度(h3)[31]。分别按式(5)、(6)计算起泡性和泡沫稳定性:

1.3.5 海藻糖脂肪酸二酯的热重分析
将10 mg的系列海藻糖脂肪酸二酯及对照物S-1170置于氮气保护的Al2O3坩埚中,使用热重分析仪分析样品,温度范围50~350 ℃,升温速率为10 ℃/min。记录在该加热过程中每个样品的质量随温度的变化值。
1.3.6 海藻糖脂肪酸二酯的细胞毒性测定
以阿霉素为对照,选取鼠巨噬RAW267.4、人高转移肝癌HCCLM3、人肝癌HepG2、宫颈癌Hela、人肺癌A549、人乳腺癌MCF-7这6种细胞系,采用噻唑蓝(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide,MTT)法评价海藻糖脂肪酸二酯的细胞毒性。6种不同的细胞加入含有10%胎牛血清的DMEM培养基培养,在37 ℃、5% CO2环境中培养12 h,移除培养基,将细胞以5 000~15 000 个/孔的密度接种到96 孔细胞培养板中,并分别与不同浓度(0、2、4、8、16、32、64、128、256 μmol/L)待测系列海藻糖脂肪酸二酯及阿霉素共同孵育24 h。分别向培养基中加入20 μL MTT溶液(5 mg/mL),37 ℃、5% CO2潮湿环境中继续孵育4 h,除去培养基,将产生的甲臜晶体溶解于200 μL二甲基亚砜中,使用酶标仪在570 nm波长处测定溶液吸光度[33]。以不添加样品及阿霉素为空白组,以样品浓度为横坐标,细胞活性抑制率为纵坐标,计算半抑制浓度(half inhibitory concentration,IC50),细胞毒性结果以IC50表示。细胞活性抑制率按式(7)计算:

式中:A实验为样品组吸光度;A空白为空白组吸光度。
1.4 数据处理与分析
采用ChemBioOffice 2019软件绘制化合物结构式;采用MestReNova 12.0软件进行化合物的谱图解析、结构确证;采用Excel 2016软件进行图表处理。采用SPSS 22.0软件进行数据统计分析,所有实验均重复3 次,结果表示为±s。采用方差分析及Duncan多重比较法进行差异显著性分析,P<0.05,差异显著。
2 结果与分析
2.1 油酸乙烯酯及海藻糖脂肪酸二酯的合成得率及结构鉴定
2.1.1 油酸乙烯酯的合成得率及结构鉴定
采用化学合成方法,经硅胶柱层析纯化反应产物,产物为透明油状液体,通过Rf值定性及NMR分析鉴定其结构即为油酸乙烯酯,经计算油酸乙烯酯的合成得率约为88%。
1H-NMR和13C-NMR鉴定结果如下:
1H NMR(300 MHz,氘代氯仿-D):δ7.28(dd,J=14.0、6.3 Hz,1H,—CH=CH2)、5.41~5.26 (m,2H,—CH=CH—)、4.86(dd,J=14.0、1.5 Hz,1H,—CH=CH2)、4.55(dd,J=6.3、1.5 Hz,1H,—CH=CH2)、2.37(t,J=7.5 Hz,2H,—CH2CO—)、2.21~1.86(m,4H,—CH2CH=CHCH2—)、1.71~1.59(m,2H,—CH2CH2CO—)、1.32~1.24(m,20H,—(CH2)4CH2CH=CHCH2(CH2)6CH3)、0.86(t,J=0.6 Hz,3H,—CH3)。加粗表示信号峰位置,下同。
13C NMR(101 MHz,氘代氯仿-D):δ170.97(C=O)、141.32(—CH=CH2)、130.14、129.83(—CH=CH—)、97.52(—CH=CH2)、34.05(—CH2CO—)、32.04(—CH2CH2CH3)、29.90、29.80、29.66、29.46(×2)、29.25、29.19、29.15(—(CH2)4CH2CH=CHCH2(CH2)4(CH2)2CH3)、27.35、27.28(—CH2CH=CHCH2—)、24.72(—CH2CH2CO—)、22.82(—CH2CH3)、14.25(—CH3)。
2.1.2 海藻糖己酸二酯
在叔丁醇-吡啶(11∶9,V/V)混合溶剂中,海藻糖在固定化脂肪酶Novozym 435催化下与己酸乙烯酯反应72 h,硅胶柱层析分离纯化,经计算海藻糖己酸二酯的合成得率约为58%。
1H-NMR、13C-NMR、MS鉴定结果如下:
1H NMR(600 MHz,氘代甲醇-D4):δ5.04(d,J=3.8 Hz,2H,H-1,H-1’)、4.36(dd,J=11.9、2.1 Hz,2H,H-6a,H-6a’)、4.20(dd,J=11.9、5.2 Hz,2H,H-6b,H-6b’)、4.01(ddd,J=10.1、5.2、2.1 Hz,2H,H-5,H-5’)、3.74~3.81(m,2H,H-3,H-3’)、3.47(dd,J=9.7、3.8 Hz,2H,H-2,H-2’)、3.34(dd,J=10.0、9.0 Hz,2H,H-4,H-4’)、2.34(t,J=7.4 Hz,4H,2×—CH2CO—)、1.66~1.59(m,4H,2×—CH2CH2CO—)、1.38~1.29(m,8H,2×—(CH2)2CH3)、0.92(t,J=7.0 Hz,6H,2×—CH3)。
13C NMR(151 MHz,氘代甲醇-D4):δ175.47(2×C=O)、95.26(C-1,C-1’)、74.54(C-3,C-3’)、73.15(C-2,C-2’)、71.88(C-5,C-5’)、71.49(C-4,C-4’)、64.36(C-6,C-6’)、35.00(2×—CH2CO—)、32.40(2×—CH2CH2CH3)、25.76(2×—CH2CH2CO—)、23.40(2×—CH2CH3)、14.28(2×—CH3)。
MS(ESI+):m/z556.3([M+NH4]+,100%)、561.3([M+Na]+,90%)。
2.1.3 海藻糖辛酸二酯
在叔丁醇-吡啶(11∶9,V/V)混合溶剂中,海藻糖在固定化脂肪酶Novozym 435催化下与辛酸乙烯酯反应72 h,硅胶柱层析分离纯化,经计算海藻糖辛酸二酯的合成得率约为45%。
1H-NMR、13C-NMR、MS鉴定结果如下:
1H NMR(600 MHz,氘代甲醇-D4):δ5.04(d,J=3.8 Hz,2H,H-1,H-1’)、4.35(dd,J=11.9、2.1 Hz,2H,H-6a,H-6a’)、4.20(dd,J=11.9、5.3 Hz,2H,H-6b,H-6b’)、4.02(ddd,J=10.1,5.2、2.1 Hz,2H,H-5,H-5’)、3.81~3.74(m,2H,H-3,H-3’)、3.47(dd,J=9.7、3.8 Hz,2H,H-2,H-2’)、3.34(dd,J=10.0、9.0 Hz,2H,H-4,H-4’)、2.34(t,J=7.4 Hz,4H,2×—CH2CO—)、1.67~1.58(m,4H,2×—CH2CH2CO—)、1.39~1.26(m,16H,2×—(CH2)4CH3)、0.91(t,J=7.1 Hz,6H,2×—CH3)。
13C NMR(151 MHz,氘代甲醇-D4):δ175.48(2×C=O)、95.25(C-1,C-1’)、74.54(C-3,C-3’)、73.15(C-2,C-2’)、71.89(C-5,C-5’)、71.47(C-4,C-4’)、64.40(C-6,C-6’)、35.05(2×—CH2CO—)、32.86(2×—CH2CH2CH3)、30.16, 30.10(2×—(CH2)2CH2CH3)、26.07(2×—CH2CH2CO—)、23.68(2×—CH2CH3)、14.43(2×—CH3)。
MS(ESI+):m/z612.3([M+NH4]+,100%)、617.3([M+Na]+,70%)。
2.1.4 海藻糖癸酸二酯
在叔丁醇-吡啶(11∶9,V/V)混合溶剂中,海藻糖在固定化脂肪酶Novozym 435催化下与癸酸乙烯酯反应72 h,硅胶柱层析分离纯化,经计算海藻糖癸酸二酯的合成得率约为75%。
1H-NMR、13C-NMR、MS鉴定结果如下:
1H NMR(600 MHz,氘代甲醇-D4):δ5.04(d,J=3.8 Hz,2H,H-1,H-1’)、4.35(dd,J=11.9、2.1 Hz,2H,H-6a,H-6a’)、4.20(dd,J=11.9、5.3 Hz,2H,H-6b,H-6b’)、4.02(ddd,J=10.1、5.3、2.1 Hz,2H,H-5,H-5’)、3.82~3.74(m,2H,H-3,H-3’)、3.47(dd,J=9.7、3.8 Hz,2H,H-2,H-2’)、3.34(dd,J=10.0、9.0 Hz,2H,H-4,H-4’)、2.34(t,J=7.4 Hz,,4H,2×—CH2CO—)、1.66~1.58(m,4H,2×—CH2CH2CO—)、1.39~1.27(m,24H,2×—(CH2)6CH3)、0.90(t,J=7.0 Hz,6H,2×—CH3)。
13C NMR(151 MHz,氘代甲醇-D4):δ175.46(2×C=O)、95.24(C-1,C-1’)、74.55(C-3,C-3’)、73.15(C-2,C-2’)、71.91(C-5,C-5’)、71.47(C-4,C-4’)、64.41(C-6,C-6’)、35.06(2×—CH2CO—)、33.05(2×—CH2CH2CH3)、30.58、30.43(×2)、30.20(2×—(CH2)4CH2CH3)、26.08(2×—CH2CH2CO—)、23.74(2×—CH2CH3)、14.46(2×—CH3)。
MS(ESI+):m/z668.4([M+NH4]+,100%)、673.4([M+Na]+,90%)。
2.1.5 海藻糖月桂酸二酯
在叔丁醇-吡啶(11∶9,V/V)混合溶剂中,海藻糖在固定化脂肪酶Novozym 435催化下与月桂酸乙烯酯反应72 h,硅胶柱层析分离纯化,经计算海藻糖月桂酸二酯的合成得率约为54%。
1H-NMR、13C-NMR、MS鉴定结果如下:
1H NMR(600 MH,氘代甲醇-D4):δ5.05(d,J=3.8 Hz,2H,H-1,H-1’)、4.36(dd,J=11.9、2.1 Hz,2H,H-6a,H-6a’)、4.20(dd,J=11.9、5.4 Hz,2H,H-6b,H-6b’)、4.01(ddd,J=10.1、5.3、2.1 Hz,2H,H-5,H-5’)、3.82~3.75(m,2H,H-3,H-3’)、3.47(dd,J=9.7、3.8 Hz,2H,H-2,H-2’)、3.34(dd,J=10.0、9.0 Hz,2H,H-4,H-4’)、2.34(t,J=7.4 Hz,4H,2×—CH2CO—)、1.66~1.57(m,4H,2×—CH2CH2CO—)、1.37~1.26(m,32H,2×—(CH2)8CH3)、0.90(t,J=7.0 Hz,6H,2×—CH3)。
13C NMR(151 MHz,氘代甲醇-D4):δ175.46(2×C=O)、95.22(C-1,C-1’)、74.55(C-3,C-3’)、73.16(C-2,C-2’)、71.92(C-5,C-5’)、71.47(C-4,C-4’)、64.42(C-6,C-6’)、35.06(2×—CH2CO—)、33.09(2×—CH2CH2CH3)、30.76、30.62、30.50、30.44(×2)、30.21(2×—(CH2)6CH2CH3)、26.08(2×—CH2CH2CO—)、23.75(2×—CH2CH3)、14.46(2×—CH3)。
MS(ESI+):m/z724.5([M+NH4]+,100%)、729.5([M+Na]+,50%)。
2.1.6 海藻糖肉豆蔻酸二酯
在叔丁醇-吡啶(11∶9,V/V)混合溶剂中,海藻糖在固定化脂肪酶Novozym 435催化下与肉豆蔻酸乙烯酯反应72 h,硅胶柱层析分离纯化,经计算海藻糖肉豆蔻酸二酯的合成得率约为62%。
1H-NMR、13C-NMR、MS鉴定结果如下:
1H NMR(600 MHz,氘代甲醇-D4):δ5.05(d,J=3.7 Hz,2H,H-1, H-1’)、4.36(dd,J=11.9、2.1 Hz,2H,H-6a,H-6a’)、4.20(dd,J=11.9、5.4 Hz,2H,H-6b,H-6b’)、4.01(ddd,J=10.1、5.3、2.1 Hz,2H,H-5,H-5’)、3.81~3.73(m,2H,H-3,H-3’)、3.47(dd,J=9.7、3.8 Hz,2H,H-2,H-2’)、3.34(dd,J=10.5,1.5 Hz,2H,H-4,H-4’)、2.34(t,J=7.4 Hz,4H,2×—CH2CO—)、1.66~1.58(m,4H,2×—CH2CH2CO—)、1.35~1.26(m,40H,2×—(CH2)10CH3)、0.90(t,J=7.0 Hz,6H,2×—CH3)。
13C NMR(151 MHz,氘代甲醇-D4):δ175.46(2×C=O)、95.20(C-1,C-1’)、74.56(C-3,C-3’)、73.16(C-2,C-2’)、71.93(C-5,C-5’)、71.48(C-4,C-4’)、64.42(C-6,C-6’)、35.08(2×—CH2CO—)、33.10(2×—CH2CH2CH3)、30.83、30.79、30.76、30.63、30.50、30.44(×2)、30.21(2×—(CH2)8CH2CH3)、26.09(2×—CH2CH2CO—)、23.75(2×—CH2CH3)、14.46(2×—CH3)。
MS(ESI+):m/z780.6([M+NH4]+,100%)、785.5([M+Na]+,50%)。
2.1.7 海藻糖棕榈酸二酯
在叔丁醇-吡啶(11∶9,V/V)混合溶剂中,海藻糖在固定化脂肪酶Novozym 435催化下与棕榈酸乙烯酯反应72 h,硅胶柱层析分离纯化,经计算海藻糖棕榈酸二酯的合成得率约为43%。
1H-NMR、13C-NMR、MS鉴定结果如下:
1H NMR(500 MHz,氘代甲醇-D4):δ5.05(d,J=3.8 Hz,2H,H-1,H-1’)、4.36(dd,J=11.8、2.3 Hz,2H,H-6a,H-6a’)、4.21(dd,J=11.9、5.4 Hz,2H,H-6b,H-6b’)、4.02(ddd,J=10.3、5.4、2.2 Hz,2H,H-5,H-5’)、3.81~3.74(m,2H,H-3,H-3’)、3.47(dd,J=9.8、3.8 Hz,2H,H-2,H-2’)、3.34(dd,J=9.5 Hz,2H,H-4,H-4’)、2.34(t,J=7.4 Hz,4H,2×—CH2CO—)、1.68~1.56(m,4H,2×—CH2CH2CO—)、1.36~1.28(m,48H,2×—(CH2)12CH3)、0.90(t,J=6.7 Hz,6H,2×—CH3)。
13C NMR(126 MHz,氘代甲醇-D4):δ175.48(2×C=O)、95.29(C-1,C-1’)、74.70(C-3,C-3’)、73.24(C-2,C-2’)、72.02(C-5,C-5’)、71.56(C-4, C-4’)、64.49(C-6,C-6’)、35.12(2×—CH2CO—)、33.03(2×—CH2CH2CH3)、30.74(×3)、30.71(×2)、30.69、30.56、30.41、30.36、30.18(2×—(CH2)10CH2CH3)、26.08(2×—CH2CH2CO—)、23.67(2×—CH2CH3)、14.36(2×—CH3)。
MS(ESI+):m/z836.7([M+NH4]+,100%)、841.5([M+Na]+,30%)。
2.1.8 海藻糖油酸二酯
在叔丁醇-吡啶(11∶9,V/V)混合溶剂中,海藻糖在固定化脂肪酶Novozym 435催化下与油酸乙烯酯反应72 h,硅胶柱层析分离纯化,经计算海藻糖油酸二酯的合成得率约为50%。
1H-NMR、13C-NMR、MS鉴定结果如下:
1H NMR(600 MHz,氘代甲醇-D4):δ5.35(t,J=4.7 Hz,4H,2×—CH=CH—)、5.05(d,J=3.8 Hz,2H,H-1,H-1’)、4.36(dd,J=11.9、2.1 Hz,2H,H-6a,H-6a’)、4.20(dd,J=11.9、5.4 Hz,2H,H-6b,H-6b’)、4.01(ddd,J=10.1、5.3、2.0 Hz,2H,H-5,H-5’)、3.81~3.69(m,2H,H-3,H-3’)、3.47(dd,J=9.7、3.8 Hz,2H,H-2,H-2’)、3.34(dd,J=9.0 Hz,2H,H-4,H-4’)、2.34(t,J=7.4 Hz,4H,2×—CH2CO—)、2.12~1.95(m,8H,2×—CH2CH=CHCH2—)、1.66~1.59(m,4H,2×—CH2CH2CO—)、1.38~1.27(m,40H,2×—(CH2)4CH2CH=CHCH2(CH2)6CH3)、0.90(t,J=7.0 Hz,6H,2×—CH3)。
13C NMR(151 MHz,氘代甲醇-D4):δ175.40(2×C=O)、130.91、130.81(—CH=CH—)、95.18(C-1,C-1’)、74.56(C-3,C-3’)、73.16(C-2,C-2’)、71.93(C-5,C-5’)、71.47(C-4,C-4’)、64.43(C-6,C-6’)、35.07(2×—CH2CO—)、33.08(2×—CH2CH2CH3)、30.87、30.83、30.64、30.47、30.37、30.32、30.21、30.20(2×—(CH2)4CH2CH=CHCH2(CH2)4(CH2)2CH3)、28.16(2×—CH2CH=CHCH2—)、26.08(2×—CH2CH2CO—)、23.76(2×—CH2CH3)、14.48(2×—CH3)。
MS(ESI+):m/z888.7([M+NH4]+,100%)、893.7([M+Na]+,30%)。
据文献报道,固定化脂肪酶Novozym 435是催化海藻糖酯合成的最优生物催化剂[6,14]。因此,本方法采用固定化脂肪酶Novozym435,在60 ℃下催化合成海藻糖脂肪酸二酯。为获得理想的得率,采用4 倍海藻糖物质的量的脂肪酸乙烯酯为酰基供体与海藻糖发生酯交换反应,并产生易从反应中除去的乙醛,使反应正向进行[27]。据Chen Jie等[29]报道,在吡啶和叔丁醇的混合溶剂中,海藻糖溶解度较高且对酶活性影响较小,因此本研究选择在吡啶和叔丁醇的混合溶剂中反应72 h。所得产物为白色无定形固体物质,且海藻糖脂肪酸二酯是唯一产物。这是因为海藻糖是对称分子,其6及6’位羟基的空间位阻小于其他位点,而固定化脂肪酶Novozym 435对海藻糖的催化位点具有选择性[27]。实验所制备系列海藻糖脂肪酸二酯的得率为43%~75%,其中海藻糖癸酸二酯的得率最高(75%),而海藻糖棕榈酸二酯的得率最低(43%),说明脂肪酸乙烯酯的碳链越长,得率越低。这可能是由于长链脂肪酸乙烯酯在混合溶剂中的溶解度较低及空间位阻较大,导致反应相对困难,从而降低了其得率[6]。
油酸乙烯酯及系列海藻糖脂肪酸二酯的结构先由TLC定性分析,再由NMR数据鉴定。其中,1H NMR确证了油酸乙烯酯分别对应于δ4.55、4.86、7.28处的乙烯基质子峰,而δ5.41~5.26处信号峰主要归因于[—CH=CH—]基团[27]。以海藻糖己酸二酯为例,其TLC定性分析的Rf值约为0.17,在1H NMR中,脂肪酸链中甲基信号的质子峰出现在δ0.92处,与羰基相连的亚甲基[—CH2(C=O)O—]信号峰出现在δ2.34处,在δ4.20、4.36处的峰归因于(H-6b,H-6b’)和(H-6a,H-6a’),在δ5.04处的信号峰对应于海藻糖部分中的(H-1,H-1’)。在13C NMR中,[—CH3]基团的化学位移为δ14.28,[—CH2—CO—]基团的化学位移为δ35.00,在δ95.26处的信号为(C1,C1’),在δ175.47处的信号可归因于羰基[—CH2(C=O)O—]。综上,确证其为海藻糖己酸二酯[6]。同理,通过TLC、1H NMR和13C NMR可确证海藻糖辛酸二酯、海藻糖癸酸二酯、海藻糖月桂酸二酯、海藻糖肉豆蔻酸二酯、海藻糖棕榈酸二酯,而海藻糖油酸二酯需再结合1H NMR中δ5.35处归因于[—CH=CH—]基团的信号峰及13C NMR中δ130.81、130.91处归因于[—CH=CH—]的信号峰进一步确证。
如图3所示,以海藻糖月桂酸二酯为代表,通过1H异核多键相关谱确认海藻糖脂肪酸二酯的酰化位置。海藻糖月桂酸二酯C-6和C-6’位的亚甲基质子与酯部分的羰基碳相互作用,从而建立酯化位点,同时允许其他关键相互作用与海藻糖和侧链亚结构相关的非羟基质子进行共振分配。

图3 海藻糖月桂酸二酯的1H异核多键相关谱图Fig.3 1H Heteronuclear multi-bond correlation spectrum of trehalose dilaurate
由MS分析结果可知,具有不同碳链长度(C6~C18)的7种海藻糖脂肪酸二酯的分子质量在正离子模式下分别在m/z556.3、612.3、668.4、724.5、780.6、836.7、888.7观察到其加合分子离子[M+NH4]+,及m/z561.3、617.3、673.4、729.5、785.5、841.5、893.7处观察到其加合分子离子[M+Na]+。这与系列海藻糖脂肪酸二酯分子质量理论值保持一致,进一步确定其为所需海藻糖脂肪酸二酯。
2.2 海藻糖脂肪酸二酯的HLB值
由图4可知,随着脂肪酸链碳原子数的增加(C6~C18),海藻糖脂肪酸二酯侧链越长,其HLB值越低。一般而言,HLB值3~6为W/O型表面活性剂,HLB值8~18为O/W型表面活性剂。本实验中系列海藻糖脂肪酸二酯HLB值为8~13,表明其可能具有作为O/W型表面活性剂应用于食品等领域的潜力。

图4 系列海藻糖脂肪酸二酯的HLB值Fig.4 HLB values of trehalose fatty acid diesters
2.3 海藻糖脂肪酸二酯的起泡性和泡沫稳定性
糖酯的发泡性能通常根据其起泡性(形成泡沫的能力)和泡沫稳定性(保持泡沫的能力)评估,受质量浓度、溶解度、脂肪酸链长和糖类型等因素影响[34]。如图5A所示,质量浓度为0.2 g/100 mL时,随碳原子数的增加,海藻糖脂肪酸二酯起泡性逐渐增加(P<0.05),海藻糖癸酸二酯的起泡性达到101.00%,海藻糖月桂酸二酯的起泡性最佳(142.67%);然而,随着碳链长度进一步延长,起泡性开始下降(P<0.05),如海藻糖棕榈酸二酯和海藻糖油酸二酯这类具有较长烷基侧链的海藻糖脂肪酸二酯几乎没有起泡性(分别为5.00%和3.67%),甚至略低于对照物(Span 20和S-1170分别为6.33%和5.67%)。值得注意的是,酰基侧链最短的海藻糖己酸二酯未表现出起泡性,这可能是由于其亲水性较高,导致表面活性降低,起泡性亦随之变差[31],这与其较高的HLB值结果一致。此外,随着碳链长度的延长,海藻糖脂肪酸二酯的分子质量增加和疏水性增强,这些含较长脂肪酸侧链的化合物在水溶液中趋于饱和,降低了其界面吸附率和向界面的传输速率,进而降低其发泡性[35]。因此,中等酰基链长的海藻糖脂肪酸二酯由因其均衡的亲水亲油性能而具有较好的起泡性。
如图5B所示,质量浓度为0.2 g/100 mL时,对照物Span 20和S-1170在20 min内均无法稳定泡沫。海藻糖癸酸二酯、海藻糖月桂酸二酯和海藻糖肉豆蔻酸二酯均显示出优于对照物的泡沫稳定性,其中海藻糖月桂酸二酯在80 min内的泡沫稳定性最佳(50%~70%)。总体而言,海藻糖脂肪酸二酯泡沫稳定性与其起泡性有关,特别是对于具有优异起泡性的海藻糖脂肪酸二酯,0.2 g/100 mL海藻糖癸酸二酯和海藻糖月桂酸二酯的起泡性分别为101.00%和142.67%,其泡沫稳定性也较强,分别为40%~60%和50%~70%。据报道[36],低分子质量表面活性剂的起泡性和泡沫稳定性取决于多种因素共同作用,包括表面活性剂在空气-水界面的吸附能力、界面流变特性和泡沫中捕获气体的扩散速率等。这些因素之间的相互作用仍需进一步研究。本实验结果表明,中等酰基链长的海藻糖脂肪酸二酯具有较好的发泡性能,可作为发泡剂应用于食品工业中。

图5 系列海藻糖脂肪酸二酯的起泡性(A)和泡沫稳定性(B)Fig.5 Foaming ability (A) and foam stability (B) of a series of trehalose fatty acid diesters
2.4 海藻糖脂肪酸二酯的热稳定性
以市售蔗糖酯S-1170为对照,对系列海藻糖脂肪酸二酯进行热重分析,结果如图6所示。对照物S-1170质量在210 ℃时急剧下降,而此时海藻糖脂肪酸二酯质量未发生变化,表明系列海藻糖脂肪酸二酯较S-1170具有更好的热稳定性。随着温度的升高,海藻糖肉豆蔻酸二酯质量下降最快,其他海藻糖脂肪酸二酯质量下降速率随着碳链长度的增加而放缓,因此海藻糖脂肪酸二酯的脂肪酸碳链长度与其热稳定性呈正相关。其中,海藻糖油酸二酯在264 ℃时质量才开始下降,当温度达到350 ℃时剩余质量为60.58%,表现出最佳热稳定性。因此,海藻糖脂肪酸二酯可作为食品乳化剂添加于需热加工操作的食品中。
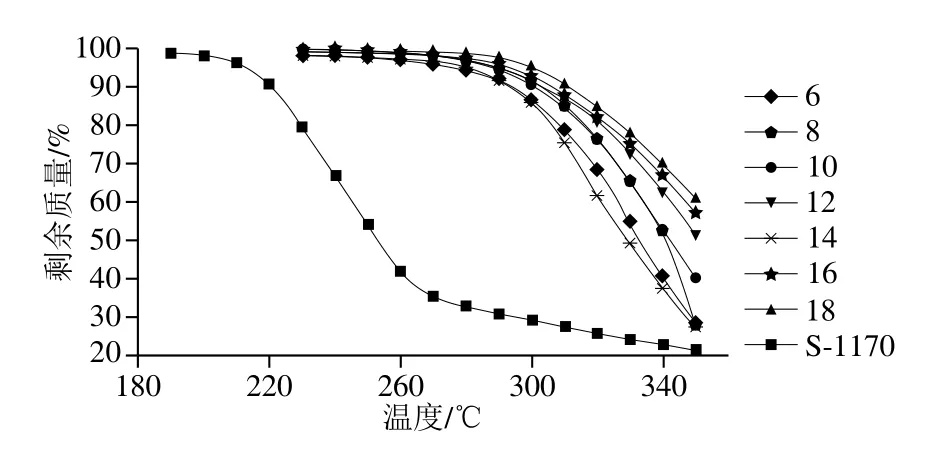
图6 系列海藻糖脂肪酸二酯的热稳定性Fig.6 Thermal stability of a series of trehalose fatty acid diesters
2.5 海藻糖脂肪酸二酯的细胞毒性
由表1可知,较低浓度的阿霉素即可使细胞死亡。相较而言,仅较高浓度的海藻糖辛酸二酯和海藻糖癸酸二酯对鼠巨噬细胞RAW267.4、人肝癌细胞HepG2和宫颈癌细胞Hela有微弱毒性。除此之外,6种细胞系在海藻糖脂肪酸二酯浓度大于256.00 μmol/L时仍能存活。这些结果说明海藻糖脂肪酸二酯普遍具有安全低毒的优点。这与Kale等[10]报道的海藻糖油酸单酯对人肝癌HepG2细胞无细胞毒性的结果一致。因此,海藻糖脂肪酸二酯或可作为安全低毒的食品添加剂添加于食品中。

表1 海藻糖脂肪酸二酯的细胞毒性Table 1 Cytotoxicity assay of trehalose fatty acid diesters
3 结 论
通过在叔丁醇-吡啶(11∶9,V/V)混合溶剂中,采用Novozym 435脂肪酶催化物质的量比为1∶4的海藻糖和系列脂肪酸乙烯酯在60 ℃下反应72 h,选择性合成6,6’-O-海藻糖脂肪酸二酯,并通过NMR谱和MS分析确证其结构,其最高得率可达75%,这为海藻糖脂肪酸二酯的合成提供了绿色有效的新方法。通过计算和测定系列海藻糖脂肪酸二酯的HLB值、发泡性能和热稳定性,发现其发泡性能随碳链长度先增大后减小,其中海藻糖癸酸二酯和海藻糖月桂酸二酯具有极好的起泡性(分别为101.00%和142.67%)和泡沫稳定性(80 min内分别为40%~60%和50%~70%)。此外,系列海藻糖脂肪酸二酯均具有较好的热稳定性,其热稳定性随碳链长度的增加而增强,其中海藻糖油酸二酯的热稳定性最好,温度达到264 ℃时质量开始下降,当温度达到350 ℃时剩余质量为60.58%,这为开发具有优越的发泡性和热稳定性的O/W型乳化剂提供了可能性。同时通过细胞毒性结果可以看出,系列海藻糖脂肪酸二酯具有安全低毒的优点。因此,海藻糖脂肪酸二酯作为一类新型糖酯,在食品工业中具有潜在的应用价值。而海藻糖脂肪酸二酯作为非离子表面活性剂的界面性质和乳化性质及其在食品中的应用需进一步研究。
猜你喜欢
生物技术进展(2022年5期)2022-10-11
中华胰腺病杂志(2022年4期)2022-08-23
新农业(2020年18期)2021-01-07
现代农村科技(2020年2期)2020-03-24
吉林农业(2019年6期)2019-06-11
教育教学论坛(2018年38期)2018-09-25
广西科技大学学报(2018年2期)2018-09-10
环球时报(2018-03-01)2018-03-01
中成药(2017年10期)2017-11-16
中国粮油学报(2017年6期)2017-07-19
