
肺炎克雷伯氏菌生物合成1,3-丙二醇的副产物调控研究
2022-01-04 09:17陶春平
化学与生物工程 2021年12期
陶春平
(常州新东化工发展有限公司,江苏 常州 213034)
1,3-丙二醇(1,3-propanediol,1,3-PD)是一种重要的化工原料,可作为有机溶剂应用于润滑油、染料、油墨、抗冻剂等行业,尤其可作为单体应用于新型聚酯材料聚对苯二甲酸丙二醇酯(PTT)的合成。PTT具有许多独特的性质:尼龙样的弹性恢复、良好的着色性、抗紫外线、抗静电等,受到研究者的广泛关注,从而促进了其合成单体1,3-PD的研究和开发[1]。为了角逐PTT的巨大市场,DuPont和Shell两家跨国公司曾采用化学合成路线,以环氧乙烷或丙烯醛为原料生产1,3-PD。但化学合成路线需要高温高压、设备投资巨大且合成的1,3-PD的羰基含量高,直接导致长期以来1,3-PD来源受限,从而限制了PTT的发展。为此,DuPont公司开发了一条生物合成路线,以谷物来源的葡萄糖为底物,通过基因工程改造的大肠杆菌发酵生产1,3-PD,在全球范围内形成了技术垄断和专利封锁[2],目前仅在美国本土生产1,3-PD,再将其作为原料出口至世界其它国家和地区。
为了打破DuPont公司的垄断,各国科学家积极寻找DuPont公司的生物合成替代路线,人们发现,自然界中存在一些细菌可以利用甘油为底物合成1,3-PD,这些细菌包括肺炎克雷伯氏菌(Klebsiellapneumoniae)、产酸克雷伯氏菌(Klebsiellaoxytoca)和丁酸梭菌(Clostridiumbutyricum)等[3];同时,随着生物柴油产业的不断发展,其副产的甘油可以为这些细菌发酵生产1,3-PD提供足够底物,因此,这种利用甘油为底物生物合成1,3-PD的路线具有较好的开发前景,得到了较多的关注。然而这些细菌在以甘油为底物合成1,3-PD的同时会产生副产物,如乙醇、乳酸、丁二酸、乙酸等,这些副产物的存在一方面降低了甘油转化率,同时还会对菌体生长产生抑制作用[4]。因此,作者选择K.pneumoniaeATCC 25955为原始菌株,利用代谢工程手段对K.pneumoniae生物合成1,3-PD的副产物进行调控,以提高K.pneumoniae生物合成1,3-PD的效率。
1 实验
1.1 菌株与质粒
K.pneumoniaeATCC 25955购于美国菌种保藏中心(American Type Culture Collection,ATCC),置于甘油管中-80 ℃冰箱保藏,该菌株具有对氧气不敏感的特性而适用于工业化生产;E.coliDH5α化学感受态细胞购于Vazyme公司,用于质粒的构建和扩增等分子克隆实验;E.coliS17-1和质粒pKR6K均由上海交通大学许平教授惠赠,E.coliS17-1用于扩增质粒pKR6K并通过接合转移的方式将其转移到K.pneumoniae中,质粒pKR6K用于目的基因的敲除和整合;质粒pDK7自带IPTG诱导型启动子tac,由英国John Innes Centre的Mike J. Merrick教授惠赠,用于在K.pneumoniae中过表达基因。实验所涉及的菌株和质粒相关信息见表 1。

表1 菌株与质粒
1.2 引物
引物(表2)用于扩增目的基因的上下游同源臂,均由金斯瑞生物科技(南京)有限公司合成。
1.3 培养基
(1)LB培养基(g·L-1):胰蛋白胨10,酵母提取物5,氯化钠10,pH值自然,121 ℃灭菌20 min。配制固体培养基时添加终浓度为20 g·L-1的琼脂。如有需要,向培养基中添加终浓度为25 μg·mL-1的氯霉素或100 μg·mL-1的氨苄。
(2)摇瓶发酵培养基(g·L-1):甘油20.0,酵母提取物7.0,K2HPO4·3H2O 7.0,KH2PO42.0,(NH4)2SO41.25,MgSO4·7H2O 0.1,微量元素1.0 mL·L-1。微量元素成分包括:CaCl2·2H2O 3.2 mg·L-1,ZnCl23.8 mg·L-1,FeCl3·6H2O 30.0 mg·L-1,MnCl2·4H2O 11.14 mg·L-1,CuCl2·2H2O 0.96 mg·L-1,CoCl2·6H2O 2.64 mg·L-1,H3BO30.35 mg·L-1,Na2MoPO4·2H2O 24.5 μg·L-1。用NaOH溶液调节初始pH值至7.0。如有需要,向培养基中添加终浓度为20 μg·mL-1的氯霉素。当OD600值达到0.6~0.8时,添加终浓度为1 mmol·L-1的IPTG。
(3)分批补料发酵培养基(g·L-1):甘油20.0,酵母提取物1.0,K2HPO4·3H2O 6.0,KH2PO42.0,(NH4)2SO45.0,MgSO4·7H2O 0.32,C6H5Na3O7·2H2O 0.6,微量元素1.0 mL·L-1。微量元素的成分同上。如有需要,向培养基中添加终浓度为20 μg·mL-1的氯霉素。当OD600值达到0.6~0.8时,添加终浓度为1 mmol·L-1的 IPTG。
(4)LAS培养基(g·L-1):蛋白胨10,酵母粉5,蔗糖150,琼脂粉15,115 ℃灭菌20 min。
(5)M9培养基(g·L-1):5×M9储存液:Na2HPO4·12H2O 8.5,KH2PO41.5,NaCl 0.25,NH4Cl 0.5;柠檬酸三钠固体培养基:柠檬酸三钠0.5,琼脂粉15;每100 mL柠檬酸三钠固体培养基中加入20 mL 5×M9储存液并加入合适浓度的抗生素。上述成分均在121 ℃下灭菌20 min。
1.4 菌种培养
摇瓶培养:将-80 ℃保藏的K.pneumoniaeATCC 25955于LB平板上划线培养得到单菌落;挑取活化的单菌落接种至50 mL LB液体培养基中,37 ℃、200 r·min-1培养12~16 h;然后将种子液转接到50 mL摇瓶发酵培养基中,37 ℃、160 r·min-1培养30 h,控制起始OD600值为0.01。
发酵罐培养:将-80 ℃保藏的K.pneumoniaeATCC 25955于LB平板上划线培养得到单菌落;挑取活化的单菌落接种至50 mL LB液体培养基中,37 ℃、200 r·min-1培养过夜;以1%的接种量转接至摇瓶发酵培养基中,37 ℃、200 r·min-1培养8~12 h;再以5%的接种量转接至5 L搅拌生物反应器(初始装液量为3 L)中,37 ℃、200 r·min-1进行分批补料发酵。发酵过程中用4 mol·L-1NaOH溶液调节pH值为7.0,通气量控制在1 vvm,当初始甘油浓度降至2 g·L-1以下时添加60%的甘油进料溶液以保持甘油浓度在20~30 g·L-1范围内。期间,控制甘油进料溶液的补充速率,确保甘油浓度每2 h增加3~5 g·L-1。
1.5 分子克隆方法
通过自杀质粒同源重组的方式实现目的基因的敲除和插入。首先,构建具有待敲除基因(Target)上下游同源臂的敲除质粒pKR6K-ΔTarget,并经过E.coliS17-1以接合转移的方式将其转移到K.pneumoniae胞内发生同源重组,从而达到敲除目的基因的目的。当在pKR6K系列敲除质粒的上下游同源臂之间插入相应目的基因后,可以相同的方式实现相应目的基因的整合表达。
在接合转移的过程中,分别吸取5 mLE.coliS17-1(pKR6K-ΔTarget)和1 mLK.pneumoniae菌液,离心并用无菌生理盐水混匀后滴至固体LB平板中间,正置37 ℃培养过夜;将平板中间菌落用无菌生理盐水吹打混匀,6 500×g离心5 min,收集菌体,并用4 mL无菌生理盐水重悬;将重悬液适当稀释后,涂布于含有氯霉素抗性的M9固体培养基上进行单交换筛选,37 ℃培养过夜;挑取M9固体培养基上的单菌落,设计引物进行菌落PCR验证并得到阳性克隆子;将阳性克隆子接种到LB液体培养基中,37 ℃、200 r·min-1培养过夜;将培养液稀释后涂布于LAS固体培养基上进行双交换筛选;最后,对目的基因进行菌落PCR验证,得到目的基因缺失的菌株。表达质粒pDK7通过电转化的方式导入K.pneumoniae中,并在IPTG诱导下进行表达[8]。
用一步组装试剂盒(ClonExpress MultiS One Step Cloning Kit,Vazyme)实现重组质粒的构建。首先选择合适的酶切位点对载体进行线性化,通过引物设计软件(http://www.vazyme.com)设计引物,在5′端加上20 bp同源序列,使扩增产物之间以及扩增产物与线性化克隆载体之间都具有能够相互同源重组的完全一致的序列。分别定量各片段浓度,并计算各片段的最适使用量(0.02×片段碱基对数)。将各片段混合吹打均匀,37 ℃反应30 min后降至4 ℃或立即置于冰上冷却。取10 μL重组产物加入到100 μLE.coliDH5α化学感受态细胞中用于重组质粒的转化。最后,设计引物进行菌落PCR,并挑取阳性克隆子提取质粒后测序验证。
1.6 检测方法
用紫外分光光度计检测菌体的生物量,以发酵液OD600值表示。发酵开始后,每隔一定时间取500 μL发酵液,10 000×g离心5 min,弃上清;用无菌生理盐水清洗2遍后重悬,稀释一定倍数后测定OD600值。菌体的细胞干重(DCW)是以上述方法收集菌体后冷冻干燥至恒重得到。
采用外标法绘制各代谢产物及底物的标准曲线。
用高效液相色谱仪(HPLC)检测主要代谢产物及底物的含量,包括乳酸、乙醇、丁二酸、1,3-PD、2,3-丁二醇、乙酸和甘油。取不同时期的发酵液于10 000×g离心5 min,上清液稀释一定倍数后经0.22 μm水相尼龙膜过滤,进样检测。色谱条件:Aminex HPX-87H色谱柱(300 mm×7.8 mm,Bio-Rad,USA),检测器为示差检测器,柱温65 ℃,流动相为5 mmol·L-1硫酸,流速0.6 mL·min-1,进样量10 μL。
2 结果与讨论
2.1 副产物合成途径缺失菌株的构建
通过设计pKR6K自杀质粒可以实现K.pneumoniae基因组中目的基因的靶向敲除,从而阻断相关代谢途径。为了有效提高1,3-PD的产量,首先,尝试敲除乙醇、乳酸、丁二酸、2,3-丁二醇等副产物合成途径中的关键基因,其关键酶的编码基因分别为adhE、ldhA、frdA、budA,大小分别为2 133 bp、990 bp、1 792 bp、780 bp。然后,分别构建能够用于基因敲除的质粒pKR6K-ΔadhE、pKR6K-ΔldhA、pKR6K-ΔfrdA、pKR6K-ΔbudA,经过测序验证,确定可以用于K.pneumoniae突变菌株的构建。将上述质粒导入E.coliS17-1中,并通过接合转移的方式使质粒在K.pneumoniae胞内与基因组上的目标片段进行同源重组,依次敲除相关基因。在得到阳性克隆子后,分别用引物adhE-F和adhE-R1进行菌落PCR扩增adhE基因,用引物ldhA-F和ldhA-R1进行菌落PCR扩增ldhA基因,用引物frdA-F和frdA-R1进行菌落PCR扩增frdA基因,将得到的阳性克隆子命名为Kpr-1。在Kpr-1的基础上,进一步对budA基因进行敲除,将得到的阳性克隆子命名为Kpr-2。
2.2 副产物合成途径缺失重组菌株的摇瓶发酵
为了验证副产物合成途径缺失对K.pneumoniae利用甘油合成1,3-PD的影响,对原始菌株WT、菌株Kpr-1、菌株Kpr-2进行摇瓶发酵,绘制生长曲线并测定主要代谢产物乳酸、乙醇、丁二酸、1,3-PD、2,3-丁二醇、乙酸的含量,结果如图1所示。
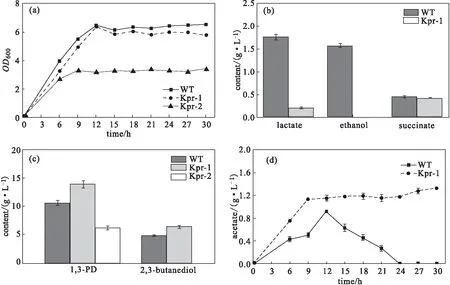
图1 原始菌株WT、菌株Kpr-1、菌株Kpr-2的摇瓶发酵生长曲线及代谢产物分布Fig.1 Growth curves and metabolite profiles of strains WT,Kpr-1,and Kpr-2 under shake fermentation
由图1可知:
(1)敲除adhE、ldhA、frdA基因后,乙醇、乳酸、丁二酸的合成途径缺失,菌株Kpr-1摇瓶发酵副产物乳酸和乙醇的积累显著减少,乳酸含量从1.76 g·L-1降至0.22 g·L-1,乙醇含量从1.58 g·L-1降至0 g·L-1,与文献[9]报道一致;但丁二酸积累变化不明显,含量仅从0.46 g·L-1降至0.43 g·L-1(图1b);目标产物1,3-PD含量在发酵末期能够达到13.92 g·L-1,相比原始菌株WT提高了31.0%(图1c);乙酸含量明显升高并呈现持续积累的趋势,在发酵末期达到1.33 g·L-1(图1d);菌株Kpr-1的生物量明显减少,仅为原始菌株的88%(图1a),这是由于质子化的乙酸可以穿过细胞膜并破坏跨膜pH梯度,从而对细胞产生毒性[10],抑制菌体生长甚至导致发酵停止。由于胞内的代谢扰动,2,3-丁二醇的含量也有所变化,从4.80 g·L-1升至6.35 g·L-1(图1c)。这是由于2,3-丁二醇的合成会受到发酵过程中pH值的诱导,在酸性条件下,2,3-丁二醇合成途径会被激活以抵抗这种逆环境;在当前发酵过程中,高浓度的乙酸积累不可避免地会导致培养基逐渐酸化,从而促进2,3-丁二醇的合成[11]。
(2)在菌株Kpr-1的基础上,进一步敲除budA基因,2,3-丁二醇合成途径缺失,虽然可以有效降低2,3-丁二醇含量,但是会严重影响菌株Kpr-2的生物量以及目标产物1,3-PD含量。菌株Kpr-2的生物量在发酵末期仅为菌株Kpr-1的60%,并且提前3 h进入稳定期(图1a);目标产物1,3-PD的含量大幅降低,从13.92 g·L-1降至5.59 g·L-1。究其原因,主要是2,3-丁二醇合成途径对于促进辅因子NADH的再生以及维持胞内pH值稳定具有重要作用。据文献[12]报道,在K.pneumoniae阻断2,3-丁二醇和乳酸合成途径的同时会不可避免地造成酸(特别是丙酮酸)的积累,从而影响菌株的发酵性能。另外,2,3-丁二醇合成途径中的中间代谢产物还参与了支链氨基酸的生成,支链氨基酸对于微生物的生长至关重要,缺乏这类物质必然会抑制菌体生长。因此,后续实验保留2,3-丁二醇合成途径,以Kpr-1作为出发菌株,进一步对乙酸溢流现象进行代谢调控,以进一步提高1,3-PD的产量和生产强度。
2.3 乙酸吸收途径的强化
菌株Kpr-1与原始菌株WT相比具有更高的1,3-PD产量和生产强度。然而,由于胞内代谢流的再分配,导致进入乙酸合成途径的碳通量增加。在K.pneumoniae发酵过程中乙酸的合成主要通过AK-PTA合成途径。然而,这条合成途径对于调控菌体胞内代谢平衡以及维持菌体生长具有重要作用,因而无法直接对其进行阻断。另一方面,通过序列比对发现K.pneumoniae发酵过程中存在一条能够回用乙酸的代谢途径,由乙酰辅酶A合成酶(ACS,由acs基因编码)催化乙酸回用生成乙酰辅酶A。为了减少乙酸的积累,作者采用强化乙酸吸收途径的代谢策略,加快乙酸的吸收,缓解乙酸溢流。研究过程中,分别对来自K.pneumoniae和巴氏醋杆菌(Acetobacterpasteurianus)的acs基因进行过表达,探究了应用上述策略对K.pneumoniae乙酸积累的影响。最后,筛选了更加高效的acs基因进行基因组整合表达,以更好地满足工业化生产需求。
2.3.1 构建乙酰辅酶A合成酶过表达菌株
为了强化乙酸吸收途径来解决K.pneumoniae发酵过程中乙酸溢流的问题,分别构建能够过表达内源乙酰辅酶A合成酶基因(acskp)的质粒pDK7-Ppc-acskp和能够异源过表达A.pasteurianus来源乙酰辅酶A合成酶基因(acsap)的质粒pDK7-Ppc-acsap。用含有氯霉素的抗性平板进行阳性克隆子的筛选,获得能够更高效催化乙酸转化为乙酰辅酶A的acs基因。经过测序验证后,将以上质粒通过电转化的方式分别转化到菌株Kpr-1中,构建重组菌株Kpr-1-Ppc-acskp、Kpr-1-Ppc-acsap。
为了将Ppc-acsap表达盒整合到菌株Kpr-1的基因组中进行组成型表达,构建质粒pKR6K-frdA::Ppc-acsap,并将其通过接合转移的方式导入菌株Kpr-1中,通过多轮抗性筛选得到阳性克隆子,将其命名为Kpr-3。
2.3.2 乙酸吸收途径强化重组菌株的摇瓶发酵
为了探究强化乙酸吸收途径对K.pneumoniae利用甘油合成1,3-PD的影响,对菌株Kpr-1、重组菌株Kpr-1-Ppc-acskp、重组菌株Kpr-1-Ppc-acsap进行摇瓶发酵,绘制生长曲线并测定底物甘油及主要代谢产物1,3-PD、乙酸、2,3-丁二醇、乳酸、乙醇、丁二酸的含量,结果如图2所示。
由图2可知:
(1)引入acs基因后,重组菌株Kpr-1-Ppc-acskp、Kpr-1-Ppc-acsap的生物量均有所升高,与菌株Kpr-1比较分别提高了13.7%和17.7%(图2a);重组菌株的生物量在发酵开始的12 h内要明显低于菌株Kpr-1。
(2)引入acs基因后,重组菌株的甘油利用速率稍有下降。发酵6 h时,菌株Kpr-1的甘油剩余量就低于2株重组菌株;发酵9 h时,就能够完全消耗培养基中的甘油;而重组菌株要在发酵12 h后才能完全消耗底物甘油(图2b)。
(3)引入acs基因后,重组菌株的乙酸含量明显下降,表现出更强的乙酸回用能力。在发酵0~9 h时,2株重组菌株积累乙酸的含量较菌株Kpr-1低;发酵9 h后,乙酸更是被持续吸收并在发酵27 h时被消耗完毕,说明强化乙酸吸收途径这个代谢工程策略能够有效调控发酵过程中乙酸的含量。从图2d可以看出,引入acsap基因的重组菌株Kpr-1-Ppc-acsap表现出更好的乙酸吸收效果。因此,选择Ppc-acsap表达盒进行基因组整合表达。随着乙酸的持续消耗,重组菌株Kpr-1-Ppc-acskp、Kpr-1-Ppc-acsap的生物量在发酵12 h后均有所提高;但2株重组菌株的1,3-PD含量在发酵末期均下降了约11%(图2c),推测目标产物1,3-PD的产量可能与乙酸的生产具有潜在的关系。
(4)引入acs基因后,2,3-丁二醇和乳酸的含量基本没有变化(图2e、f),碳代谢流在这两条合成途径之间保持着一种总量平衡。
(5)值得注意的是,乙酰辅酶A衍生物的产量受到了过表达acs基因的影响,直观的表现为发酵过程中乙醇和丁二酸的含量提高了(图2g、h)。总之,强化乙酸吸收途径能够有效促进乙酸回用,减少发酵过程中乙酸的积累。

图2 菌株Kpr-1、重组菌株Kpr-1-Ppc-acskp、重组菌株Kpr-1-Ppc-acsap的摇瓶发酵生长曲线及代谢产物分布Fig.2 Growth curves and metabolite profiles of strains Kpr-1,Kpr-1-Ppc-acskp,and Kpr-1-Ppc-acsap under shake fermentation
2.3.3 乙酸吸收途径强化重组菌株的分批补料发酵
考虑到后续的产物分离难度及经济成本,在发酵过程中应避免使用抗生素(用于维持质粒稳定)、化学诱导剂(例如IPTG)等价格昂贵的额外添加剂[13]。将组成型启动子pc控制的Ppc-acsap表达盒整合到基因组中进行表达能够有效解决上述问题。为了进一步探究重组菌株Kpr-3的发酵性能,对该菌株进行分批补料发酵并对主要代谢产物含量进行测定,结果如图3所示。
由图3可知,在分批补料发酵条件下,菌株Kpr-3的1,3-PD产量显著提升,达到94.9 g·L-1,生产强度达到3.16 g·L-1·h-1。值得注意的是,在发酵末期,乙酸含量相较于菌株Kpr-1下降了17.6%,从10.8 g·L-1降至8.9 g·L-1,表明上述乙酸溢流调控策略可以有效调节K.pneumoniae发酵过程中乙酸的积累。然而,菌株Kpr-3在发酵中期(10~18 h)产生了较多的乙酸,可以推断acs基因的连续表达不可避免地增加了乙酰辅酶A的代谢通量,从而激活乙酸合成途径,为细胞生长提供更多能量,以补偿乙酰辅酶A合成酶消耗的ATP。实际上,内源性acs基因的表达可以响应乙酸合成途径的通量增加和碳限制,从而使其代谢通量上调[14]。当底物碳被持续消耗时,acs基因开始表达,与此同时乙酸开始被重新回用。随着发酵的进行,分批补料条件下甘油的不断添加直接导致底物过剩情况出现,此时乙酸含量又开始提高。根据理论计算,乙酸合成途径对1,3-PD的合成更为有利[15]。当乙酸是甘油代谢的唯一副产物时,可获得合成1,3-PD的最大NADH供应量[16]。因此,在此阶段(10~18 h),1,3-PD产量明显提高,并在发酵20 h后达到平稳。相似的结论在丁酸梭菌中也有发现,即1,3-PD的高浓度与有机酸(乙酸和丁酸)的高浓度有关[17]。另外,强化乙酸吸收途径对乳酸含量的影响几乎可以忽略不计。与此同时,2,3-丁二醇的含量下降了13.4%,为29.0 g·L-1。丁二酸和乙醇的最终含量分别为5.0 g·L-1和0.49 g·L-1,与菌株Kpr-1相似,但菌株Kpr-3在发酵中期的丁二酸含量明显提高,推测这是由于乙酰辅酶A代谢通量增加所致。总之,乙酰辅酶A合成酶的整合表达可以有效提高1,3-PD的最终产量。

图3 菌株Kpr-1(a)和重组菌株Kpr-3(b)分批补料发酵的代谢产物分布Fig.3 Metabolite profiles of strains Kpr-1(a) and Kpr-3(b) under fed-batch fermentation
3 结论
利用接合转移的方法,依次敲除了K.pneumoniae代谢甘油产1,3-PD的副产物乙醇、乳酸、丁二酸合成途径中的关键酶编码基因后,在分批补料发酵条件下,1,3-PD的产量达到83.8 g·L-1,生产强度提至2.79 g·L-1·h-1,底物甘油的转化率能够达到0.60 mol/mol,分别提高了9.07%、8.94%及13.46%。然而,胞内代谢流再分配导致副产物乙酸的积累大幅增加,从4.3 g·L-1增至10.8 g·L-1。为了减少发酵过程中的乙酸积累,进一步构建了乙酸吸收途径强化重组菌株。通过筛选比对不同来源的乙酰辅酶A合成酶基因(acs)发现A.pasteurianus来源的acsap基因能够更高效地催化乙酸合成乙酰辅酶A。将该基因进行基因组整合表达后发现,与未进行乙酸回用强化菌株相比,在分批补料发酵条件下,1,3-PD的产量和生产强度分别提高了13.24%和13.26%,达到94.9 g·L-1和3.16 g·L-1·h-1;乙酸含量从10.8 g·L-1降至8.9 g·L-1,下降了17.6%。这表明通过敲除副产物合成途径关键酶编码基因结合乙酸吸收途径强化能够促进1,3-PD的合成以及减少副产物的产生,提高K.pneumoniae代谢甘油合成1,3-PD的产量和生产强度。
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
中老年保健(2022年3期)2022-08-24
成都医学院学报(2022年4期)2022-08-19
中学生数理化·高一版(2022年4期)2022-05-09
中学化学(2021年11期)2021-12-09
江西农业学报(2021年4期)2021-04-20
中华养生保健(2020年9期)2021-01-18
三农资讯半月报(2020年11期)2020-06-21
中学科技(2017年11期)2017-12-26
食品界(2016年10期)2016-09-10
