
结外型Rosai-Dorfman病7例临床病理分析
2021-12-23 06:41庞丽娟张海俊董双双梁伟华蒋金芳
临床与实验病理学杂志 2021年11期
冯 肖,庞丽娟,张海俊,王 宁,董双双,梁伟华,蒋金芳,齐 妍
Rosai-Dorfman病(Rosai-Dorfman disease, RDD)又称伴巨淋巴结病的增生症(sinus hisliocytosis with massive lymphadenopathy, SHML),是一种伴炎症和窦组织细胞增生的慢性病变,主要表现为非朗格汉斯窦组织细胞增多[1],其发病年龄14.8~51.4岁[2]。2016年组织细胞学协会将RDD划分为组织细胞的“R组”,皮肤RDD被单独划分为组织细胞的“C组”[3]。RDD临床表现多样,主要分为淋巴结型、结外型和混合型[4]。RDD镜下组织学形态多样,典型特点是结内淋巴窦广泛扩张和大量淋巴细胞、浆细胞和组织细胞浸润,同时组织细胞吞噬淋巴细胞、红细胞和浆细胞[4]。RDD与郎格汉斯组织细胞增生症(Langenhans cell histiocytosis, LCH)、IgG4相关疾病相似,临床易误诊。本文着重探讨RDD的临床病理学特征、诊断、鉴别诊断,为临床与病理医师提供参考。
1 材料与方法
1.1 材料收集2012年1月~2020年9月石河子大学第一附属医院诊断的7例RDD患者,其中女性5例,男性2例,发病年龄32~63岁,病程8个月~2年,治疗方式主要为手术切除。
1.2 方法标本均经10%中性福尔马林固定,常规脱水,石蜡包埋,3 μm厚切片,常规HE染色,镜下观察。免疫组化染色采用EnVision法。抗体包括S-100、CD1a、CD68、IgG、IgG4、CD34、CK(AE1/AE3)、desmin、SMA、vimentin、ALK、CD21、Ki-67,均购自北京中杉金桥公司。应用Leica BOND-MAX全自动免疫组化仪进行染色。
2 结果
2.1 临床特点7例患者中4例因肿物包块就诊,1例因面部肿痛就诊,1例因鼻塞就诊,1例因面部皮疹就诊。发病部位:3例位于头颈部、2例位于小腿皮下组织、2例位于乳腺。6例患者有影像学检查:1例CT示右侧鼻前庭、鼻中隔前缘区有一小结节占位,相邻结构受累,考虑肿瘤可能(图1);1例B超示双侧颌下区低回声,诊断为颌骨肿物;1例B超示皮下软组织增厚水肿并异常低回声;3例B超分别示皮下、皮下脂肪层及皮下软组织内异常回声。
2.2 病理检查
2.2.1眼观 7例肿物为灰红、灰黄色不整形组织(图2),直径1.3~6.0 cm;6例无包膜,1例有包膜;6例与周围组织界限稍清楚,1例与周围组织界限欠清。
2.2.2镜检 7例均有组织细胞增生,多量淋巴细胞、浆细胞等浸润,病变可见明区和暗区。明区低倍镜下见增生的纤维组织和苍白的组织细胞,高倍镜下组织细胞呈泡沫样;暗区可见大量淋巴细胞和浆细胞聚集(图3)。部分病例以明区为主,间质纤维组织增多及血管周围炎细胞浸润,伴随小灶及片状凝固性坏死,高倍镜下见杆状核类上皮样细胞及大圆形核组织细胞。部分病例以暗区为主,低倍镜下有较多淋巴细胞、浆细胞围绕血管呈靶环样(图4);7例中仅有1例高倍镜下见组织细胞胞质内吞噬数量不等浆细胞的组织学特征(图5)。
2.3 免疫表型病变中组织细胞S-100呈胞质阳性(图6),CD68阳性(图7),CD1a(图8)阴性。2例CD34血管阳性,2例SMA部分阳性;2例IgG4阳性,IgG均阴性,故未评估IgG4/IgG阳性比;desmin、vimentin、ALK、CD21均阴性;Ki-67增殖指数为5%~35%,提示部分区细胞增殖较活跃。
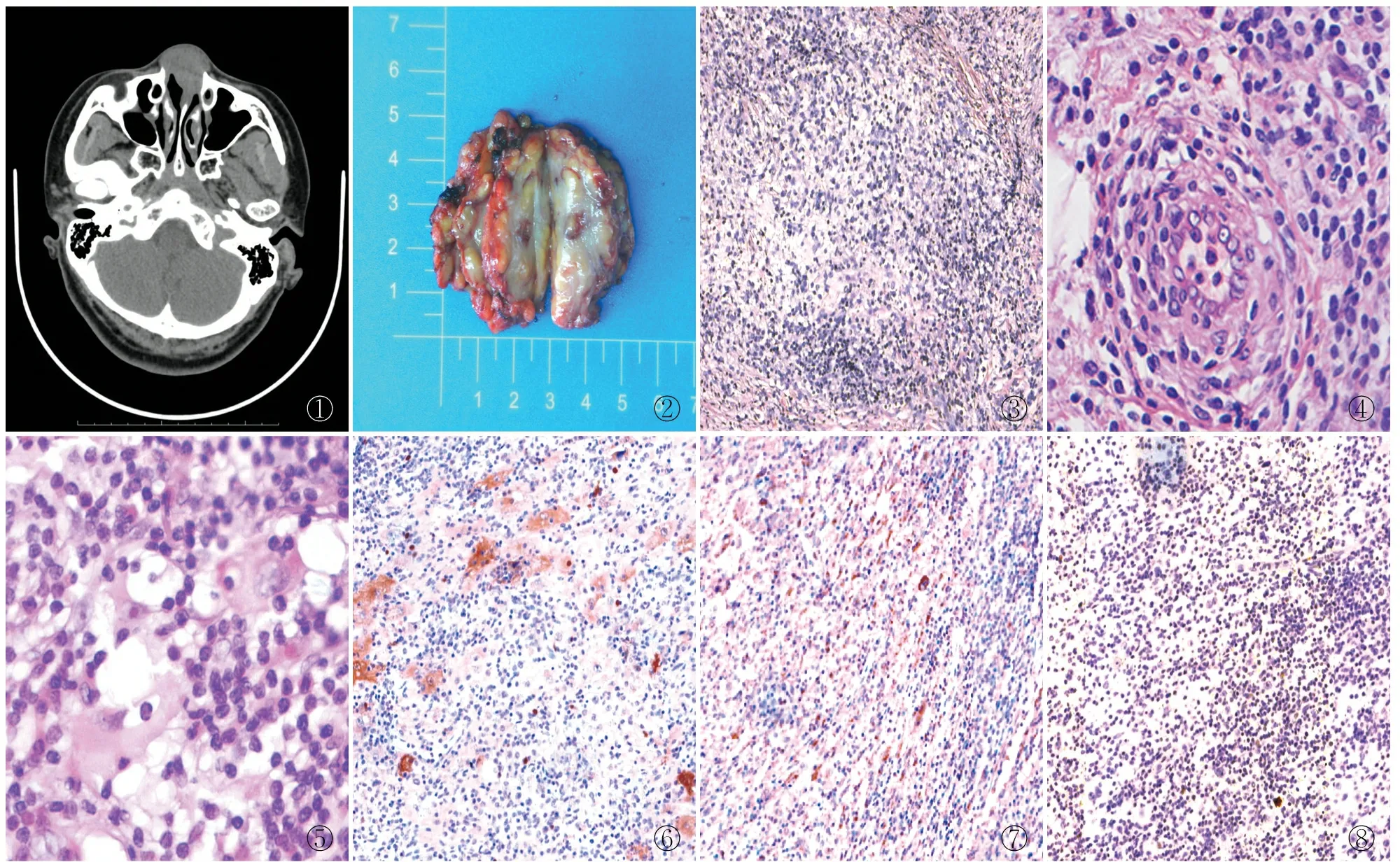
①②③④⑤⑥⑦⑧
2.4 随访7例患者中有2例存活且复发,3例存活无复发,2例失访。
3 讨论
1965年RDD首次被Destombes报道,1969年由Rosai和Dorfman命名[5]。约30%的患者伴发热[6],少数患者出现盗汗和体重减轻等症状[3]。目前,RDD的病因尚不清楚,有文献报道其可能与遗传、感染和炎症有关[7]。RDD临床表现多种多样,结外型RDD有大量的纤维组织增生时易掩盖其典型组织学形态而误诊为LCH、结核和IgG4相关疾病[8]。RDD多数情况下可自愈,当病变累及器官[9],尤其累及肾脏和肺时,预后较差。
3.1 临床特点RDD男女发病比为1.4 ∶1,发病年龄14.8~51.4岁[2]。RDD以淋巴结型常见,结外型次之,混合型罕见,结外型主要发生于头颈部[10]。患者可出现发热、盗汗和体重减轻症状,有些患者出现白细胞升高、血沉增快和丙球蛋白升高等现象,当病变累及肺、肾脏等器官时出现相应肿块,进而导致呼吸和泌尿系统等并发症。
3.2 病理特征结内型RDD主要特征:(1)淋巴结结构破坏,淋巴窦广泛扩张,窦内充满淋巴细胞、浆细胞、组织细胞以及形态多样的组织细胞和小单核细胞。组织细胞和淋巴细胞构成明暗区[11]。(2)组织细胞较大,胞质丰富嗜酸性,偶呈泡沫状,胞核形态多样[3]。(3)组织细胞可吞噬完整的淋巴细胞、中性粒细胞和红细胞,该特征称为吞噬现象或伸入运动[8]。结外型RDD的组织学特征与结内型RDD类似,但组织细胞较少,伸入运动不常见[12],同时纤维化现象较多。本组7例RDD均为结外型,均伴不同程度的纤维化,仅1例有伸入运动。
3.3 免疫表型和分子遗传学特征免疫组化作为诊断RDD的必要检测手段,其诊断依据主要为组织细胞S-100、CD68阳性和CD1a阴性[1];也可出现CD163阳性,CD34、CD21阴性;T 细胞CD4、CD8、CD45阳性;B细胞CD20、CD79a阳性;特征性的组织细胞vimentin、CD11c和MAC387可阳性[8]。在分子遗传学方面,RDD发病机制可能与KRAS、MAP2K突变、人类疱疹病毒及细菌感染有关[13]。有报道发现艾滋病毒感染者可同时患有RDD[11],其可能与病毒、细菌感染及免疫系统缺陷有关。
3.4 鉴别诊断(1)LCH:具有组织细胞增多并伴炎症特征,包括朗格汉斯细胞、单个核细胞和树突状细胞[14],LCH无伸入运动,Langerin阳性和CD1a阴性可初步排除LCH[3]。(2)间变性大细胞淋巴瘤:典型特征是淋巴窦和血管周围浸润大量淋巴细胞、浆细胞和肿瘤细胞,与结内型RDD的组织学特征极其相似。但其肿瘤细胞呈多核、异型核以及CD30阳性特征[15]。(3)IgG4相关疾病:属于罕见性疾病,主要特征是肿块样硬化性病变,部分病例有过度纤维化,与RDD组织学特征较相似。RDD可伴随IgG4和IgG阳性的浆细胞,RDD诊断共识建议IgG4/IgG阳性比值>40%作为诊断IgG4相关疾病的临界点[16]。(4)慢性炎性病变:组织学上以淋巴细胞为主的慢性炎性改变,大量淋巴细胞、浆细胞浸润。可见纤维母细胞和增生的小血管,偶见组织细胞呈空泡状,但体积比RDD的组织细胞小。该病亦不会出现异型核、多核组织细胞,组织细胞胞质S-100阴性[17]。
3.5 治疗与预后目前,RDD患者的治疗原则是采取保守观察、定期随访。RDD的预后相对较好,有报道显示超过50%的患者可自行缓解,但也有约10%的患者死于RDD累及重要脏器而导致的并发症和感染[3]。当RDD累及重要脏器或形成较大肿块时,需进行手术切除,术后再辅以抗菌或化疗药物,这是常见且有效的治疗手段。皮肤RDD服用激素类药物可有效缓解症状。皮质类固醇疗法、化疗和免疫疗法,适用于全身RDD患者[9]。然而,当RDD患者出现KRAS和MAP2K的突变,以上治疗方式均无效,需行分子靶向治疗[13]。现阶段RDD的治疗方式尚无统一标准,应进行早发现、早治疗、综合个体化治疗。
猜你喜欢
首都食品与医药(2022年12期)2022-12-06
中国临床医学(2022年3期)2022-07-08
临床超声医学杂志(2022年4期)2022-05-07
现代消化及介入诊疗(2021年9期)2021-11-23
医学信息(2021年5期)2021-03-21
发明与创新·中学生(2018年11期)2018-11-30
上海医药(2016年6期)2016-04-28
安徽医科大学学报(2015年9期)2015-12-16
企业导报(2015年16期)2015-12-14
浙江中医杂志(2004年9期)2004-03-08
