
4-(2,6,6-三甲基-3-羟基-1-环己烯-1-基)-3-丁烯-2-酮的微波纯化研究 *
2021-11-23 03:52邵开元胡文祥
湖北科技学院学报 2021年5期
李 冉,闵 清,邵开元,胡文祥
(1.湖北科技学院 药学院,湖北 咸宁 437100;2.北京神剑天军医学科学院 京东祥鹄微波化学联合实验室,北京 101601)
微波是频率在 300 MHz 到 300 GHz 范围内、波长在 1 mm~1 m 之间的电磁波[1,2]。而微波化学是一门新兴的前沿交叉科学,是利用现代微波技术来研究物质在微波场作用下的物理和化学行为的一门科学,常用于加速化学反应或者催化新反应的进行,对于提高反应效率有重要作用。本文主要是对虾青素的重要中间产物4-(2,6,6-三甲基-3-羟基-1-环己烯-1-基)-3-丁烯-2-酮进行纯化,采用微波化学技术加速反应的进行,并结合正交试验得到最佳工艺条件。
虾青素是一个多烯化合物,具有多种生理功能,其强大的抗氧化能力越来越受到人们的重视。虾青素的来源主要包括天然提取和人工合成,而目前,虾青素天然来源是通过雨生红球藻的提取分离获取,由于提取率低下,也难以满足人们日益增长的需求量,故通过全合成方法获得虾青素也逐渐被重视起来。人工合成虾青素,由于其反应步骤较长,目前有多种合成路线。本文采用的是以α-紫罗兰酮为起始原料的合成路线[3,4]。其反应式如下图1所示:
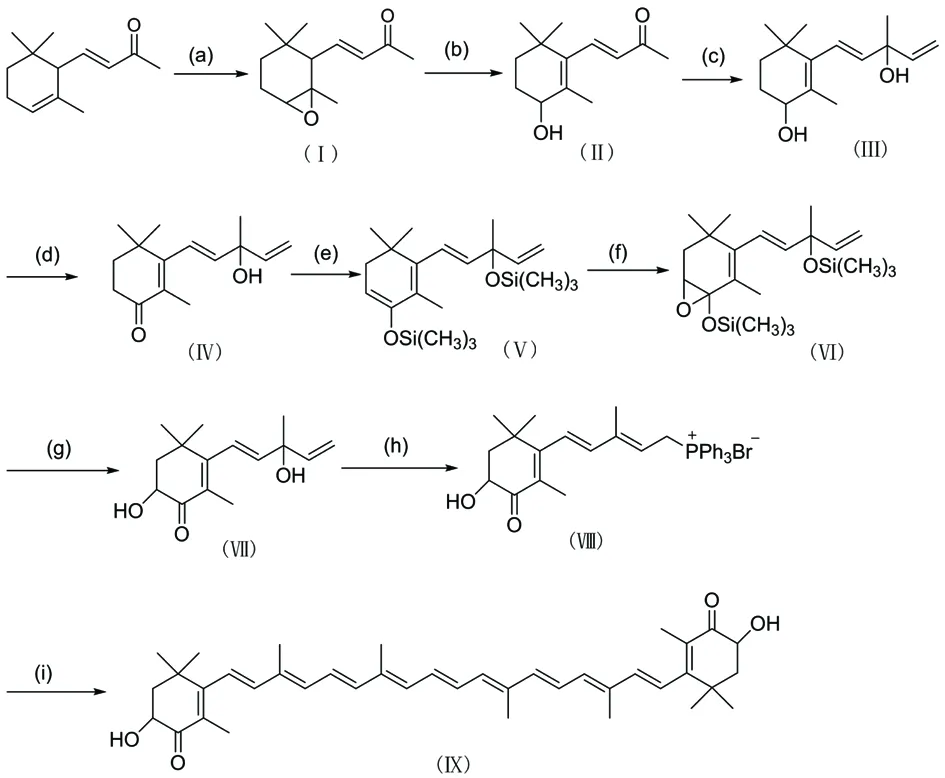
图1 虾青素的合成路线
(a) m-CPBA/CH2Cl2, 0 ℃;
(b) NaOCH3/CH3OH;
(c) CH2=CHMgCl/THF, -30 ℃ ;
(d) Aluminum isopropoxide/acetone, reflux;
(e) LDA/TMSCl;
(f) m-CPBA/CH2Cl2, -20℃;
(g) K2CO3/CH3OH, reflux;
(h) HBr/PPh3;
(i) NaOCH3/CH3OH,2,7-dimethyl-2,4,6-octantrien-1,8-dial, 0 ℃.
在合成虾青素的过程中,中间体的合成是其关键步骤。一些重要中间体产物如羟基酮(Ⅱ),其纯度越高,后续副产物越少,越有利于反应的进行。然而中间体(Ⅱ)往往呈现非固体非液体的黏稠状,很难依靠传统的重结晶和蒸馏方法提纯。羟基酮(Ⅱ)的结构是有含两个烯烃、一个羟基和一个羰基的不饱和羟酮化合物,用常规方法难以纯化。皮士卿等人采用其特殊官能团结构用化学法进行提纯。其过程为图2:

图2 普通方法提纯
因为化合物(Ⅱ)含有一个羟基,可与琥珀酸酐反应,产物是一个有机酸的中间体[5]。而主要杂质成分为二酮化合物和单酮化合物,分别为下图3和图4化合物:
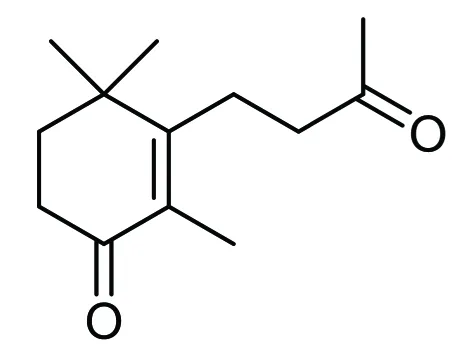
这两个化合物均不含羟基,不与琥珀酸酐反应。这样,反应产物用碱处理后,可将这两个非酸的杂质弃掉,酸化后即可得到较纯的有机酸化合物,然后用甲醇钠处理,即回到化合物(Ⅱ)。
然而,该提纯过程中重要的是合成的化合物(Ⅱ)与琥珀酸酐反应。按照文献报道的操作方法,用乙酸乙酯作溶剂,5个小时回流,但效率低下。
通过研究发现,在反应中,反应时间越长,产生的副产物也会逐渐提高。而微波工艺具有缩短反应时间,提高反应效率的作用,故本文应用微波法和酰化催化剂相结合,用无水甲苯作溶剂且进行正交试验,优选最佳的实验方案。来进行反应的纯化工艺的探索。
一、正交试验设计与结果
1.正交试验设计
由于纯化过程中使用了微波技术,而微波反应原料配比,反应时间,微波功率对反应结果会产生不同的影响,为了得到最好的反应效果,本实验采用了正交试验设计来探索其反应的最佳工艺[6,7]。
本实验考查了原料间配比(A)、反应时间(B)、微波功率(C)三个因素在三水平上纯化效率的影响。具体实验操作为:分别取不同配比的粗品和琥珀酸酐于250mL三口瓶中,加入少量酰化催化剂DMAP(4-二甲氨基吡啶), 搅拌均匀后,在XH-300UL电脑微波超声波紫外光组合催化合成仪中按不同水平进行反应,以薄层色谱分析来进行判断反应终点。然后用定量的色谱甲醇溶解,并用高效液相色谱分析(外标法)。
色谱分析柱:迪马C18 (250 × 4.6 mm, 5 μm);流动相:乙腈-水,紫外检测波长:410 nm;流速:1 ml/min;柱温为30℃,采集时间为15 min。
按照正交表进行,因素水平表见表1,正交试验结果表见表2。
原料1:丁二酸酐 原料2:粗品酮(Ⅱ) 配比为:原料1:原料2
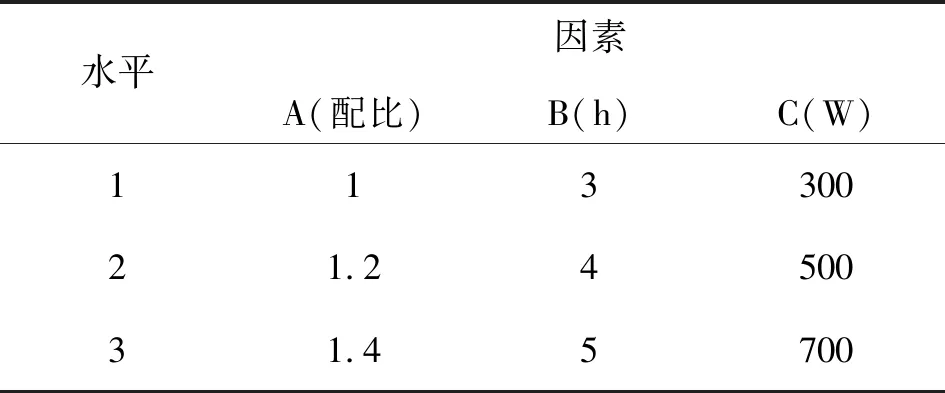
表1 纯化粗品酮(Ⅱ)实验的因素水平表
2.正交试验结果
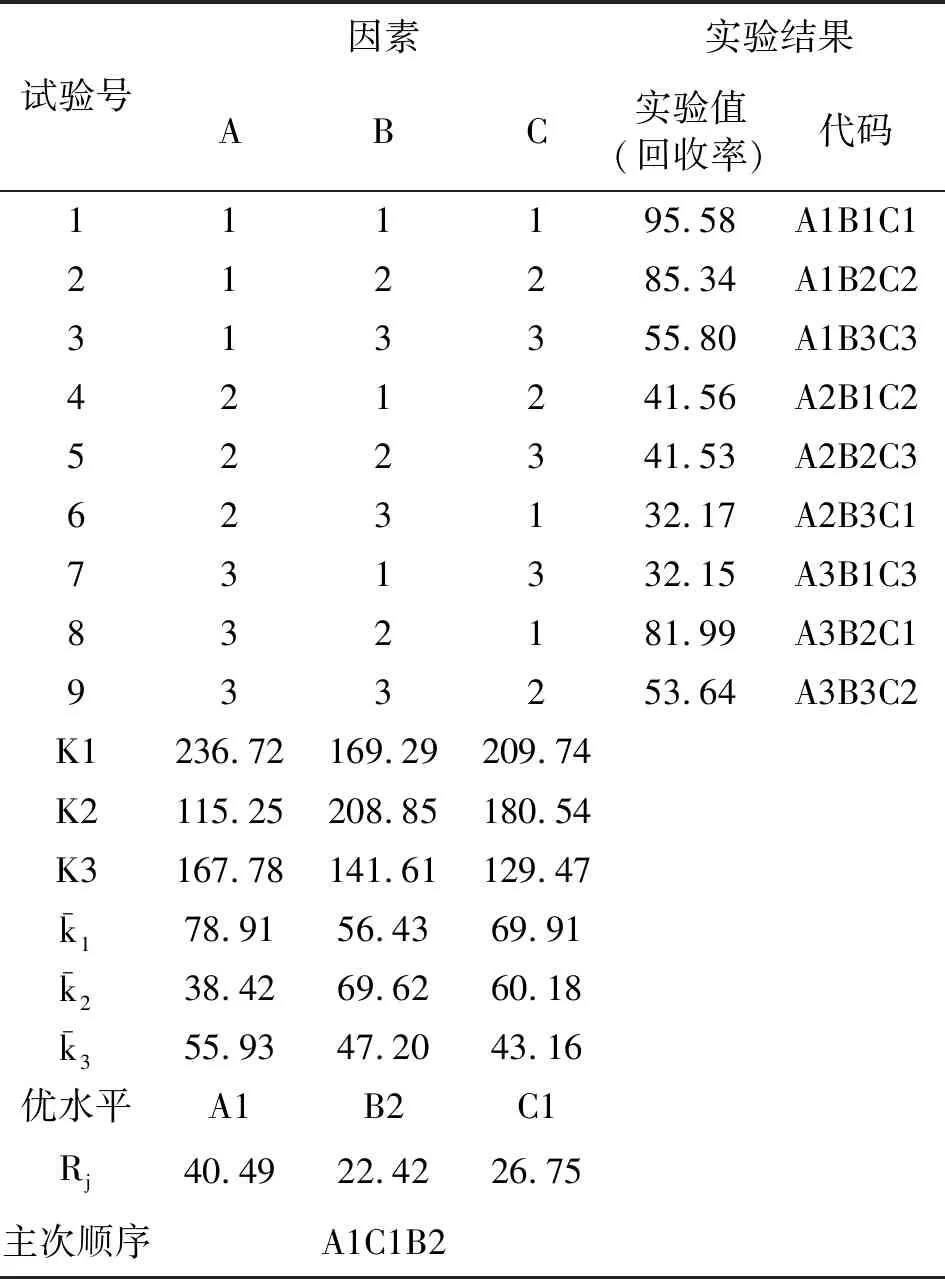
表2 纯化粗品酮(Ⅱ)正交试验结果
由上述正交试验可知,纯化中间体产物粗品酮(Ⅱ)的微波反应条件为原料配比为1∶1,微波功率300W,反应时间4h,且优先顺序为配比、功率、时间。在上述试验范围内可得到最佳的纯化工艺。
二、实验部分
1.仪器与试剂
仪器:XH-200A电脑微波固液相合成/萃取仪,XH-8000Plus多用途微波化学合成仪。
主要试剂:丁二酸酐,甲苯,甲醇钠,DMAP(4-二甲氨基吡啶)
2.实验步骤
反应路线如下图5所示
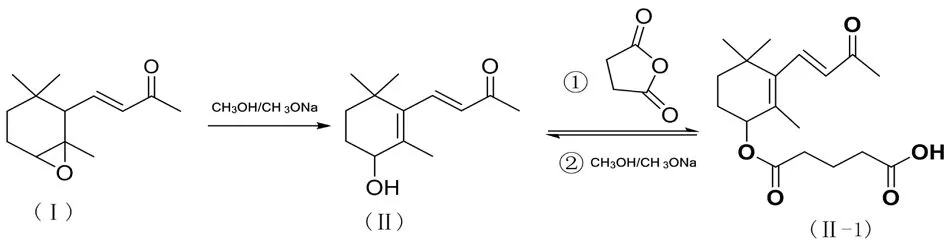
图5 化合物Ⅱ的纯化路线
将环氧物Ⅰ溶于320mL的无水甲醇中,加入20mL甲醇钠溶液(30%),回流反应5h,薄层色谱测定反应完全后,冷却至室温,用15mL冰醋酸中和至pH为7后,将甲醇溶液减压回收,加水80mL,用乙酸乙酯萃取3次,再用饱和食盐水洗涤一次,合并有机层,用无水硫酸镁干燥后,过滤,滤液即为羟酮化合物Ⅱ的粗品。
粗品需要经过两步反应进行进一步的纯化,纯化过程为将上述过滤的萃取液蒸除溶剂,加入甲苯溶解后,将溶液转移至500mL三颈瓶中,之后加入琥珀酸酐(44.0g,0.4mol)和少量酰化催化剂DMAP(4-二甲氨基吡啶),将反应体系置于微波固液相合成仪中,微波反应回流4h,功率300W,温度120℃,薄层色谱鉴定反应完全后,冷却至室温,反应液用饱和碳酸钠水溶液萃取,萃取液pH为9后,分液漏斗分液,弃去有机层,水层用大量20%HCl酸化至pH为2,再用乙酸乙酯萃取3次,合并有机层后,有无水硫酸镁干燥过夜,过滤减压蒸除溶剂,得到羟酮化合物的六元环羟基被琥珀酸酐保护的中间体产物[8,9]。将反应中间体加入200mL甲醇,再加入30%的甲醇钠溶液(68.0g 0.42mol),微波反应2h,薄层色谱显示反应终点后,将反应液冷却至室温,减压除去溶剂后,加入100mL蒸馏水,用乙酸乙酯萃取3次,合并有机层,干燥后过滤蒸除溶剂,即得纯羟酮化合物Ⅱ。
三、结果与讨论
1.结果
本研究是微波技术在有机合成中的成功应用。微波催化反应避免了高压高温的危险性,将反应能迅速地催化完成,极大提高了反应效率,提高反应的安全性,催化剂的加入更是加快了反应的进行。本次纯化也结合了正交试验方法,获得最优实验方案[10]。得到最佳数据为原料配比为1∶1,微波功率300W,反应时间4h,且优先顺序为配比、功率、时间,而研究结果表明,本研究方法可以获得高纯度的羟基酮产物,并对其进行核磁结果的确认,谱图结果与实验结果一致。且本方法操作简便,效率高。对微波化学的拓展应用提供了新的领域[11~13]。其核磁结果为:
1H NMR (400 MHz, Chloroform-d) δ 7.20 (dt, J = 16.5, 1.1 Hz, 1H), 6.13 (d, J = 16.5 Hz, 1H), 5.31 (s, 1H), 4.03 (t, J = 5.0 Hz, 1H), 2.32 (s, 3H), 1.94 (dddd, J = 14.5, 11.5, 5.4, 3.5 Hz, 1H), 1.86 (s, 3H), 1.80 - 1.62 (m, 2H), 1.47 (ddd, J = 13.4, 7.7, 3.4 Hz, 1H), 1.07 (d, J = 11.8 Hz, 6H)。
2.讨论
随着微波化学的进一步发展,微波催化反应理论研究也离不开其热力学及动力学的研究,建立微波催化反应理论也是新时代的呼唤[14]。而微波技术的应用已经被广泛应用到各个领域中,例如,微波合成,高分子材料分解等,近年来更是应用到环境科学领域,如污水治理工程,环境监测与分析等方面已取得巨大进展[15]。本文采用的微波纯化工艺和方法尚未在文献报道,可推广应用到其他化合物的提纯及一些中药材的提取等工艺研究中。对于化合物的提纯工艺可提供一定的理论依据。
猜你喜欢
中南民族大学学报(自然科学版)(2022年4期)2022-07-01
中国药学药品知识仓库(2022年10期)2022-05-29
中国饲料(2021年17期)2021-11-02
科学与财富(2021年33期)2021-05-10
落叶果树(2021年6期)2021-02-12
汽车零部件(2018年5期)2018-06-13
山东工业技术(2018年9期)2018-05-26
股市动态分析(2015年12期)2015-09-10
中小企业管理与科技·中旬刊(2014年10期)2015-02-03
建材发展导向(2014年2期)2014-05-04
