
细胞自噬与肾脏疾病研究中国专家共识
2021-11-10 09:13中国病理生理学会肾脏病专业委员会刘华锋杨俊伟
中国病理生理杂志 2021年10期
中国病理生理学会肾脏病专业委员会; 刘华锋, 杨俊伟
(1广东医科大学附属医院肾病研究所,广东湛江 524001;2南京医科大学第二附属医院肾脏病中心,江苏南京 210003)
1963 年比利时生物化学家Duve 第一次使用自噬(autophagy)这一名称,他同时因发现溶酶体获得了1974 年的诺贝尔生理学或医学奖。日本科学家大隅良典因发现了细胞自噬机制而荣获2016 年诺贝尔生理学或医学奖。目前认为细胞自噬是一种溶酶体依赖的细胞降解途径。自噬包括生理状态下的基础自噬以及应激状态下所诱导的自噬[1],在肾脏固有细胞的生理及病理过程中均发挥重要作用。目前我国肾脏病领域的同道对细胞自噬基本理论知识的认识参差不齐,对自噬检测方法的合理使用需进一步规范,特别是未来如何以细胞自噬为切入点开展各种肾脏疾病发病机制研究有待进一步明确。为此,中国病理生理学会肾脏病专业委员会(Chinese Association of Pathophysiology,Society of Nephrology,CAPN)组织相关专家通过查阅Medline、Biomedicine和MEDLINE Search 有关自噬文献1 000 余篇,参考2021 年国际Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy,结合中国专家在自噬领域,特别是自噬与肾脏病研究的经验,首次制定了这部专家共识。共识介绍了自噬分类、形成机制、检测技术及自噬失衡与各种肾脏病发病机制的关系,并总结靶向自噬通路相关分子防治肾脏疾病新进展和未来趋势,希望为广大从事肾脏病临床与基础研究的工作者提供有益参考。
1 细胞自噬分类
根据待降解的底物类型和向溶酶体运送底物的途径,自噬可分为微自噬(microautophagy)、分子伴侣介导的自噬(chaperone-mediated autophagy)和巨自噬(macroautophagy)。前两者直接发生在溶酶体上,不涉及双层膜结构的自噬小体(autophagosome)。其中,微自噬是通过溶酶体膜的内陷主动、直接吞噬胞浆成分[2];分子伴侣介导的自噬则是通过热休克同源蛋白70(heat shock cognate protein 70,Hsc70)特异性识别含有KFERQ五肽蛋白成分,通过溶酶体相关膜蛋白2(lysosomal-associated membrane protein 2,LAMP2)和其它辅因子的多聚体将待降解的物质传递到溶酶体内进行降解[3];而巨自噬需形成大小为0.5~2 μm 的双层膜结构自噬小体[4]。巨自噬是细胞自噬中最常见的自噬类型,故一般将其简称为“自噬”。
另外,根据底物是否具有选择性,细胞自噬(巨自噬)又可分为非选择性自噬和选择性自噬。当营养匮乏时,非选择性自噬激活,底物随机被包裹进自噬泡进行降解;而细胞出现特异性细胞器损伤时,则一般启动选择性自噬,包括线粒体自噬、溶酶体自噬、核糖体自噬、细胞核自噬及内质网自噬等[5]。选择性自噬的分子机制各不相同,比如溶酶体严重损伤后膜通透性增加,其内β-半乳糖苷快速被胞浆中的半乳糖凝集素3(galectin-3;作为感应器)识别并结合,使受损的溶酶体被特异性的标识,结合到溶酶体膜上的galectin-3 招募TRIM16(tripartite motif-containing 16;作为适配器)并募集E3 泛素连接酶,促使溶酶体膜蛋白泛素化,继而使ATG16L1(autophagyrelated 16-like 1)和ULK1(Unc-51-like kinase 1)等自噬起始因子激活,溶酶体被特异性包裹进自噬泡进而被降解[6]。因此,选择性自噬研究重点集中在寻找特异性的感受器和适配器,以及相关调节机制。

专家推荐意见一:1. 根据待降解的底物类型和向溶酶体运送底物的途径,推荐自噬分为微自噬、分子伴侣介导的自噬和巨自噬3 类进行相关研究。2. 根据底物是否具有选择性,建议将细胞自噬分为非选择性自噬和选择性自噬进行研究。
2 关于“自噬流”
“自噬流”(autophagic flux)是指细胞自噬发生的全过程,包括起始、成核、延伸、成熟、融合和降解以及降解产物转运至细胞质(图1)[1]。在自噬起始阶段,丝氨酸/苏氨酸蛋白激酶ULK1、ULK2和其它结合蛋白形成的复合体发挥重要作用[7];而磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)复合物调节囊泡成核和自噬泡形成[8];ATG12-ATG5-ATG16L系统和微管相关蛋白1 轻链3(microtubule-associated protein 1 light chain 3,MAP1LC3;又称LC3)系统则控制自噬囊泡扩张和最终自噬小体形成[9]。因此,在研究细胞自噬时应该特别注意“自噬流”形成过程中的不同关键分子,并深入开展相关机制研究。其次,自噬体与溶酶体融合形成自噬溶酶体(autolysosome)是自噬的后续阶段,被自噬小体捕获的物质被运输到溶酶体进行降解,所产生的生物小分子通过溶酶体内特异性受体运输至细胞浆重新利用。细胞骨架相关的运动蛋白、可溶性N-乙基马来酰亚胺敏感因子附着蛋白受体(solubleN-ethylmaleimide-sensitive factor attachment protein receptor,SNARE)等在调控自噬溶酶体形成过程中可能发挥重要作用[10],但其机制还不十分清楚,因此也是一个值得研究的课题。此外,最近研究提示自噬溶酶体重构(autophagic lysosome reformation,ALR)促进溶酶体新生,但其病理生理学意义及相关机制也值得深入探讨[11]。

专家推荐意见二:1. 推荐研究“自噬流”形成过程中的不同关键分子及相关机制。2. 建议深入探讨自噬溶酶体形成及自噬溶酶体重构的机制和关键调控分子。
3 细胞自噬的调控
细胞自噬可响应多种因素而启动,如活性氧簇(reactive oxygen species,ROS)、内质网应激、缺氧、DNA 损伤和免疫信号等,但其活性的高低受到启动转录及转录后蛋白修饰调控,其中哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)通路、AMP 活化蛋白激酶(AMP-activated protein kinase,AMPK)通路、乙酰化修饰等发挥关键作用(图1)。这些调控因素会激活或抑制自噬,使细胞能够快速适应或抵抗瞬时细胞应激,以维持细胞生命活动。
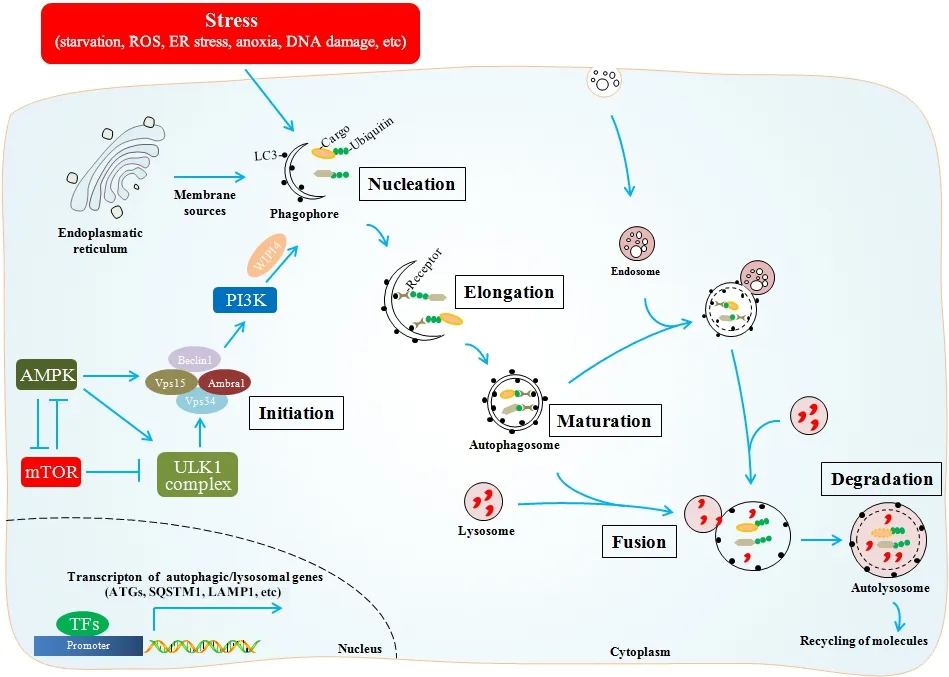
Figure 1. Schematic presentation depicts the autophagic flux and main regulatory mechanisms(provided by Institute of Nephrology,Affiliated Hospital of Guangdong Medical University). The autophagic flux is divided into initiation,nucleation,elongation,autophagosome maturation,fusion with lysosomes,and degradation of autophagic cargoes. Autophagy is regulated by transcription factors,kinases(mTOR,AMPK,etc)and post-translational modifications.图1 自噬流的过程及主要分子调控机制(广东医科大学附属医院肾病研究所供图)
3.1 转录水平调控 自噬转录调控是指通过促进自噬相关基因或溶酶体相关基因的表达调节细胞自噬活性,其中,针对小眼症转录因子E(microphthalmia/transcription factor E,MiT/TFE)家族的转录因子EB(transcription factor EB,TFEB)蛋白研究最为突出。已知TFEB 主要调节溶酶体生物发生、溶酶体胞吐、脂肪吞噬等。在营养丰富或非应激条件下,TFEB 处于非活性状态;当细胞应激时,TFEB 被去磷酸化激活,发生核转位,启动自噬相关基因的表达,促进溶酶体新生并与自噬体融合,从而促进自噬通量。另外,核受体超家族的一些转录因子如Rev-Erbα/NR1D1(nuclear receptor subfamily 1,group D,member 1)以组织特异性方式调节自噬相关基因表达。如NR1D1 通过直接与DNA 序列中的调控区结合,抑制骨骼肌中Ulk1、Bnip3(Bcl-2/adenovirus E1B 19 kD-interacting protein 3)、Atg5和Becn1(beclin-1)等基因的表达[12];而在肌肉组织中,NR1D1上调自噬相关基因表达(如Ulk1等),促进自噬。此外,叉头框蛋白O(forkhead box protein O,FOXO)家族的转录因子也参与多个自噬相关基因的转录[13]。其次,表观遗传相关蛋白如乙酰转移酶TIPO60可通过促进组蛋白H4的乙酰化诱导自噬基因表达,进而促进细胞自噬。其它如亚精胺、褪黑素、液泡膜蛋白1(vacuole membrane protein 1,VMP1)、早期生长反应蛋白1(early growth response protein 1,EGR1)等都可参与自噬相关基因的转录调控,从而调节自噬通量[14]。目前,上述分子在肾脏病自噬研究领域仍有较大的研究空间。
3.2 mTOR通路 mTOR是一种进化保守的丝氨酸/苏氨酸蛋白激酶,是磷脂酰肌醇相关蛋白激酶家族的成员。大量研究表明,mTOR是自噬的关键调节因子。mTOR 参与形成2 种类型的复合物,即雷帕霉素敏感的mTOR 复合物1(mTOR complex 1,mTORC1)和不敏感的mTORC2。其中,mTORC1信号是自噬发生过程的“守门员”,它通过与下游信号分子真核翻译起始因子4E结合蛋白1(eukaryotic translation initiation factor 4E-binding protein 1,4E-BP1)、核糖体蛋白S6 激酶1(S6 kinase 1,S6K1)相互作用,启动并调控自噬相关基因的转录和翻译,参与自噬水平的调节[15]。在营养丰富时,mTORC1 通过介导ULK1(Ser637 和Ser757)和ATG13(Ser258)特定位点磷酸化,抑制ULK1 复合物的活性,从而抑制自噬小体的生物发生;而在饥饿和细胞应激时,mTORC1 活性被抑制而与ULK1 分离;游离的ULK1 通过Thr180 磷酸化而激活募集并磷酸化ATG13、FIP200(focal adhesion kinase family-interacting protein of 200 kD)、ATG101 和其他ATG 蛋白形成ULK1 复合物,随后转移到内质网的隔离膜上,启动自噬[16]。mTORC1也可通过磷酸化ATG14、AMBRA1 和NRBF2(nuclear receptor binding factor 2)直接调节Ⅲ型磷脂酰肌醇3-激酶复合物1(class Ⅲphosphatidylinositol 3-kinase complex 1,PI3KC3-C1)的活性,抑制自噬的成核步骤[17-18]。此外,mTORC1 还可以通过靶向WIPI2(WD repeat domain phosphoinositide-interacting protein 2)和p300 乙酰转移酶参与调节自噬体形成的延伸步骤:如通过磷酸化p300,促进LC3 乙酰化,阻碍LC3脂化,从而抑制自噬[19]。进一步,mTORC1 磷酸化WIPI2并阻碍其与ATG16L的结合,从而抑制ATG12-ATG5-ATG16L 复合物结合到自噬体膜上,干预磷脂酰乙醇胺对LC3脂化的促进作用,最终抑制自噬[20]。
3.3 AMPK 通路 AMPK 是一种异源三聚体蛋白质激酶,也是诱导自噬的重要因素。AMPK 主要通过2种信号调控自噬:mTOR依赖和非依赖两种方式。在细胞营养缺乏时AMPK 被激活[21],通过抑制mTORC1 复合物的形成及活化,激活ULK1 复合物,启动细胞自噬。AMPK 还可通过磷酸化ULK1 而直接启动细胞自噬[22]。此外,AMPK 还可以直接介导beclin-1 的Thr388 位点磷酸化,促使beclin-1 与Bcl-2解离,促进PI3KC3-CI的形成,产生大量磷脂酰肌醇-3-磷酸(phosphatidylinositol-3-phosphate,PI3P),从而促使自噬泡生成[23]。AMPK 还可以促进ATG16 复合物的形成,募集ATG5-ATG12 催化LC3 的羧基末端甘氨酸共价连接到吞噬泡膜而形成自噬泡[24],从而促进自噬进程。相反,在葡萄糖充足的情况下,活化的mTORC1 通过磷酸化ULK1 特定位点(Ser757)破坏ULK1 与AMPK 之间的相互作用而阻止ULK1 活化,从而抑制自噬启动。
3.4 脱乙酰酶sirtuins(SIRTs)通路 SIRTs 是一类参与代谢调节的NAD+依赖性Ⅲ类组蛋白脱乙酰酶,包括SIRT1 到SIRT7 共7 种蛋白[25]。它们都具有高度保守的NAD+结合域和催化功能域,不同的N 端和C 端可使它们能够结合不同的底物。SIRTs 蛋白家族调节多种蛋白的乙酰化修饰和ADP 核糖基修饰。在细胞能量耗竭的情况下,NAD+水平增加并激活下游SIRT1。SIRT1 直接介导转录因子FOXO1 和FOXO3的去乙酰化,所有这些都参与激活细胞自噬,以恢复细胞能量稳态[26]。SIRT1 可触发肝激酶B1(liver kinase B1,LKB1)去乙酰化而激活AMPK、降低LC3乙酰化水平来激活自噬[27]。此外,SIRT1还可以通过调控关键自噬蛋白(beclin-1、ATG5、ATG7 和LC3)赖氨酸残基去乙酰化作用而促进自噬,该激活是独立于mTOR/ULK1之外的自噬调控通路[28]。
目前,上述自噬途径蛋白及相关通路在肾脏细胞自噬中已有报道,但在不同的肾脏生理病理状态下,其确切作用与机制仍有待深入探讨,并有可能成为肾脏疾病防治的潜在靶点。

专家推荐意见三:1. 建议从基因转录、转录后蛋白修饰不同层面对肾脏细胞自噬发生的调控机制进行深入研究。2. 推荐重点探讨自噬途径相关调节分子及通路在肾脏固有细胞病理生理过程中的作用与机制。
4 细胞自噬检测
4.1 形态学观察 迄今为止,透射电镜进行形态学观察依然是自噬检测的最直接、最经典的方法,也被认为是观测自噬的金标准方法(图2)[29]。但该技术只是一种静态观察[30],也存在样本制备复杂、要求观察人员对自噬溶酶体的鉴定必须经验丰富等一些限制因素。

Figure 2. Autophagic vacuoles identified by transmission electron microscopy(TEM)[29]. Renal biopsy specimens were detected by TEM,and autophagosome(AP)and autolysosome(AL)were marked. The scale bar=50 μm.图2 透射电镜下观察到的自噬结构
4.2 自噬关键蛋白的检测 ATG 是细胞自噬的重要标志蛋白。不同ATG代表自噬通路进程的不同阶段。自噬激活后形成的LC3-II 水平能很好反映自噬体(包括自噬小体和自噬溶酶体)的数量。目前主要采用免疫印迹/免疫荧光技术检测LC3-II蛋白水平和分布。表达GFP(green fluorescent protein)-LC3[31]、mRFP(monomeric red fluorescent protein)-LC3[32]等荧光重组蛋白可用于直接观察体内外细胞自噬水平。其次,SQSTM1(sequestosome-1)/p62 是哺乳动物中首个被鉴定的自噬受体,可反映自噬过程中自噬降解情况[33]。另外,beclin-1 作为自噬起始阶段的关键蛋白也是检测自噬活性的关键指标,且有报道beclin-1-GFP 可用于观察是否形成自噬斑点[34]。此外,一些比较重要的自噬通路蛋白复合物,如ULK1复合物[35]、ATG12-ATG5-ATG16L 复合物[9]、PIK3C3/VPS34 复合物[36]等均在细胞自噬研究中广泛应用,可利用免疫荧光对上述复合物进行共定位观察,也可通过免疫共沉淀检测复合物的形成。
4.3 自噬流的检测 自噬是一动态过程,观察自噬流的动态过程可以了解自噬的强弱。目前最广泛应用的是GFP-mRFP-LC3 串联荧光蛋白[37]。在细胞中表达GFP-mRFP-LC3 后,自噬小体因有GFP 和mRFP双荧光而显黄色;当自噬小体进入溶酶体时,LC3 上的GFP由于溶酶体的酸性环境而淬灭,仅显示红色荧光。红色荧光相对增多表明自噬活性增强;黄色荧光增多,红色荧光相对较少则表明自噬受阻,此时即使自噬泡很多,自噬的强度也是减弱的(图3)[38]。此外,研究者又相继构建GFP-LC3-RFP-LC3ΔG 串联蛋白[39]、mTagRFP-mWasabi-LC3[40]等。这些自噬标记蛋白不仅可通过显微镜观察,还可利用流式细胞术等进行高通量、多参数检测[41-42]。类似地,Rosella 生物传感器也可作为替代方法[43]。其他检测方法还包括:LC3 周转(turnover)实验,它利用野生型和G120A 突变型海肾萤光素酶-LC3融合蛋白分别对自噬降解途径敏感和抵抗的特性,通过萤光素酶活性比值反应自噬流[44];p62/beclin-1比值也可反应自噬流高(比值减小)低(比值增大)[45]。这些自噬流的检测方法为我们研究肾组织细胞自噬提供了良好的工具。
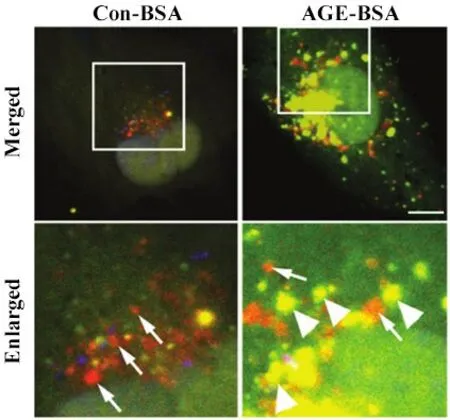
Figure 3. Advanced glycation end products(AGEs)induced autophagic blockage as monitored using tandem GFPmRFP-LC3 fusion protein[38]. HK-2 cells transfected with GFP-mRFP-LC3 plasmid were treated with AGEBSA or Con-BSA for 24 h. The yellow puncta indicate autophagosomes(arrowheads),while red puncta indicate autolysosomes(arrows). The scale bar=10 μm.图3 GFP-mRFP-LC3 串联荧光蛋白显示晚期糖基化终产物阻滞自噬流
4.4 溶酶体功能的检测 由于溶酶体是自噬通路的最终节点,溶酶体功能检测是反映自噬活性的重要方法。目前包括:(1)透射电镜形态学检查;(2)内吞体(endosome)特征蛋白Rab7 和溶酶体标志物LAMP1的抗体双标记,以检测初级溶酶体(Rab7-/LAMP1+)和次级溶酶体(Rab7+/LAMP1+)生成;(3)溶酶体组织蛋白酶检测;(4)LAMP 与组织蛋白酶或LC3 共标记;(5)利用DQ-卵清蛋白荧光淬灭实验检测溶酶体消化功能;(6)吖啶橙染色或示踪红检测溶酶体酸性环境。从现有的研究结果看,很多肾脏疾病细胞自噬异常发生于自噬通路的后端,因此,溶酶体功能检测在肾脏细胞自噬的研究中尤为重要,值得重视。
4.5 选择性自噬的检测 目前在肾脏疾病研究领域较多关注的是线粒体自噬和溶酶体自噬,其具体方法如下:
4.5.1 线粒体自噬检测 包括:(1)透射电镜及免疫电镜观察;(2)线粒体自噬相关标志蛋白(TOM20、VDAC、SOD2等),结合LC3-II或者LAMP1/LAMP2等进行免疫荧光、免疫组化、免疫印记等方法检测(图4);(3)新近开发的mt-Keima 荧光蛋白探针,可体内外检测线粒体自噬[46];(4)特异性靶向线粒体的串联荧光蛋白mCherry-GFP-FIS1[47]检测;(5)线粒体特异性探针MitoTracker 结合溶酶体抑制剂羟氯喹也可用于检测线粒体自噬流(mitophagic flux)[48]。

Figure 4. Chloroquine(CQ)suppressed mitophagy as detected by double immunofluorescent staining using TOM20 and LC3 antibodies(provided by Institute of Nephrology,Affiliated Hospital of Guangdong Medical University,unpublished data). HK-2 cells were treated with CQ(8 μmol/L)for 24 h. Afterwards,immunofluorescence microscopy was employed to detect the co-localization of outer mitochondrial membrane protein TOM20 and autophagosomes(LC3). CQ inhibited lysosomal activity,thus further impairing depletion of mitochondria via mitophagy. Consequently,mitochondria were accumulated,and increased co-localization of TOM20 and LC3 was observed(arrowheads). The scale bar=5 μm.图4 TOM20和LC3免疫荧光染色检测氯喹抑制线粒体自噬(广东医科大学附属医院肾病研究所提供,未发表数据)
4.5.2 溶酶体自噬检测 检测溶酶体自噬通常利用galectin-3 能识别受损的内吞体这一特性,构建GFP 和mRFP 串联表达的galectin-3 荧光蛋白(tandem fluorescent-tagged galectin-3,tfGal3)来检测溶酶体自噬,黄色斑点表示受损溶酶体[49](图5)。此外,也可通过免疫荧光标记galectin-3、LAMP1 和LC3 检测溶酶体自噬[50]。
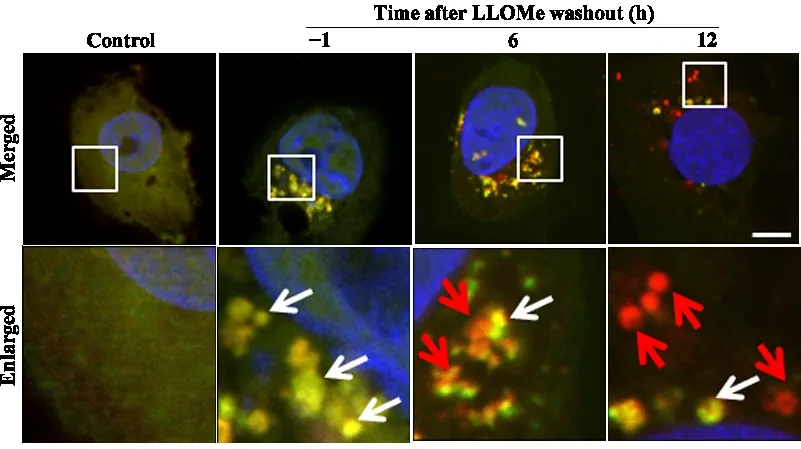
Figure 5. L-leucyl-L-leucine methyl ester(LLOMe)-induced lysophagy was detected using mRFP-GFP tandem fluorescent-tagged galectin-3(tfGal3). HK-2 cells were transfected with tfGal3. LLOMe(200 μmol/L)was added for 1 h,prior to refreshing the culture medium. Subsequently,fluorescence was monitored at 6 h and 12 h after LLOMe washout. Yellow puncta(white arrowheads)were observed at the beginning of lysosome damage. As lysophagy processed,red fluorescence(red arrowhead)maintained,while GFP was quenched in acidic lysosomes. The scale bar=10 μm. The images were provided by Institute of Nephrology,Affiliated Hospital of Guangdong Medical University,unpublished data.图5 tfGal3检测LLOMe导致的溶酶体自噬(广东医科大学附属医院肾病研究所提供,未发表数据)
值得注意的是,由于细胞自噬的动态性、敏感性和复杂性等,研究者必须结合多种检测方法才能准确判断自噬水平的高低。其中最有效的手段是前述的检测技术联合应用自噬流抑制剂,如wortmannin、3-甲基腺嘌呤、氯喹、巴弗洛霉素A1、亮肽素、E64d及胃蛋白酶抑制剂A 等[51]。目前它们是公认的评估自噬活性最常见和经济的方法,值得研究者重视。

专家推荐意见四:1. 强烈推荐以透射电镜观察作为自噬检测的经典方法。2. 推荐采用免疫印迹/免疫荧光技术检测自噬关键蛋白表达水平。3. 推荐采用GFP-LC3 等重组蛋白动态观察体内外细胞自噬水平。4. 溶酶体是自噬通路的终点,建议在自噬研究中检测溶酶体功能。5. 推荐采用mt-Keima、mCherry-GFP-FIS1 等荧光蛋白研究线粒体自噬。6. 推荐采用tfGal3荧光探针检测溶酶体自噬。
5 细胞自噬与肾脏发育、生理及衰老
5.1 自噬与肾脏发育和生理 肾脏固有细胞自噬缺陷的小鼠均能存活,且出生时肾脏结构和功能与野生型小鼠基本一样[52],提示细胞自噬在肾脏发育过程中并不发挥重要作用。但自噬在维持出生后小鼠的肾脏生理功能中发挥重要的作用。肾上皮细胞(SIX2 阳性)Atg5敲除的自噬缺陷小鼠,出生后2 月龄时出现白蛋白尿,至4 月龄时出现典型的局灶节段性肾小球硬化症[53]。特异性敲除足细胞Atg5的小鼠在出生2~4 月龄时肾小球超微结构以及尿白蛋白水平均与野生型小鼠相近,并未发生明显变化,但8~12 月龄时,敲除鼠出现足突融合和微量白蛋白尿。另外,特异性敲除肾小球内皮细胞Atg5的小鼠,出生后10 周龄时出现肾小球毛细血管轻度扩张、毛细血管袢内皮窗孔消失以及足细胞足突融合[54],补充抗氧化剂治疗,可显著缓解肾小球内皮细胞自噬缺陷引起的肾小球病变[55]。利用他莫昔芬诱导小鼠肾小管上皮细胞时间点特异性自噬缺陷,给药抑制小管细胞自噬体形成并引起p62 和泛素化蛋白堆积,5 个月开始出现肾功能损伤[52]。近端肾小管上皮细胞特异性自噬缺陷的小鼠在8 周龄时,尿葡萄糖和氨基酸排泌增加,提示肾小管功能出现障碍[56]。
在生理状态下,肾脏细胞借助基础自噬维持细胞稳态。成年肾脏固有细胞中足细胞基础自噬水平最高,而小管系统基础自噬水平较低[31,52]。小鼠禁食后,所有肾脏细胞自噬体和自噬溶酶体显著增加,说明自噬是肾脏固有细胞对营养缺乏的重要保护机制。另外,肾脏细胞选择性自噬水平和细胞总体自噬水平并不完全一致,利用mito-QC 转基因小鼠观察发现肾皮质近端肾小管细胞线粒体自噬活性最强,而皮髓交接部S3段肾小管上皮细胞线粒体自噬则较低[47]。
以上研究证实细胞自噬对维持生理状态下足细胞、内皮细胞和近端肾小管上皮细胞功能非常重要。但到目前为止,细胞自噬在系膜细胞和肾小球壁层上皮细胞中的作用尚未被揭示,因此需要我们重视。
5.2 自噬与肾脏衰老 细胞衰老的特征是细胞进入不可逆的细胞周期停滞,细胞内蛋白合成、修饰或降解等异常,并出现衰老相关分泌表型(senescence-associated secretory phenotype,SASP),后者通过分泌炎症因子和生长因子影响周围细胞而促进脏器炎症和纤维化,加速脏器衰老失功[57]。细胞自噬活性的降低与肾脏固有细胞衰老密切相关。抑制自噬体形成或者阻碍自噬体降解导致自噬缺陷,进而引起足细胞早衰,表现为大量受损线粒体无法清除、氧化修饰和泛素化修饰蛋白大量堆积,足细胞丢失及肾小球硬化,最终引起蛋白尿和肾功能衰竭[58]。与之类似,特异性敲除小管细胞Atg5的小鼠的小管细胞出现p62 和泛素化蛋白堆积、细胞DNA损伤、线粒体DNA损伤以及线粒体功能障碍,小管萎缩和间质纤维化等衰老表型[52]。目前细胞自噬对肾脏细胞衰老的作用机制尚未完全阐明,可能与线粒体稳态、细胞周期阻滞、细胞外基质分泌异常及炎症小体活化等有关。此外,调节SIRT1、AMPK、mTOR及Klotho等表达,增加端粒酶活性及限制卡路里摄入均可通过维持细胞自噬平衡,减轻肾脏细胞衰老[59]。总之,随着年龄增加,细胞自噬在衰老中的作用更为重要,进一步阐明细胞自噬对肾脏衰老的作用与机制将有重要意义。

专家推荐意见五:1. 细胞自噬在肾脏发育及维持足细胞、内皮细胞和小管上皮细胞稳态方面发挥重要作用,但在系膜细胞、肾小球壁层上皮细胞等中的作用与机制尚不十分清楚,建议加强此方面的研究。2. 细胞周期与衰老相关,细胞自噬在肾脏细胞衰老中的作用与机制尚未完全阐明,建议进一步加强细胞自噬对细胞周期和肾脏衰老的作用与机制研究。
6 细胞自噬与肾脏疾病
6.1 急性肾损伤(acute kidney injury,AKI) 在缺血再灌注引起的AKI中,近端肾小管上皮细胞自噬被激活;肾近端小管特异性敲除Atg5或Atg7基因则可加重肾缺血再灌注所引发的AKI,说明自噬激活在近端小管急性损伤过程中发挥保护作用[56]。在顺铂所致AKI 中,近端肾小管上皮细胞自噬诱导早于caspase 激活和细胞凋亡,抑制自噬加重AKI,表明自噬激活也起保护作用[60];在脓毒血症AKI也有类似的观察结果[61]。此外,线粒体自噬在缺血、肾毒性物质及脓毒性血症引起的AKI 中也被证实起保护作用。另外,AKI 除了发生小管上皮细胞变性坏死,还表现为间质炎性改变,持续的炎症反应不但加重AKI 病情,也是AKI 向慢性肾脏病(chronic kidney disease,CKD)转化的促进因素。AKI 状态下,自噬具有抑制肾间质炎症反应的作用。其主要调节机制包括:(1)AKI 发生后机体所产生的病原相关分子模式(pathogen-associated molecular patterns,PAMPs)可通过调节mTOR 和AMPK 通路激活自噬而抑制炎症小体的活化;(2)自噬通过NF-κB途径调控炎症反应,促进损伤肾组织的修复[62];(3)线粒体自噬减少肾小管上皮细胞NLRP3炎性小体的表达而减轻肾间质炎症[63]。
6.2 AKI-CKD 转化及肾脏纤维化 细胞自噬参与AKI 发生后的修复及肾脏纤维化过程(即AKI-CKD转化)。在AKI 早期,上调肾小管上皮细胞的自噬活性有助于提高其抗损伤与修复能力,延缓AKI-CKD的转化[64]。另外,自噬参与肾纤维化进展中的氧化应激、细胞凋亡、炎症、成纤维细胞增殖和活化及上皮-间充质转分化等过程。但目前细胞自噬对肾纤维化的作用仍未明确。一方面,自噬可通过维持正常溶酶体功能、线粒体自噬、细胞周期、抗凋亡[54]、抑制炎症通路、减少促炎应激产物以及介导HIF-α/FOXO3 通路[65]等起到抗纤维化作用。另一方面,自噬通过促进受损细胞持续存活[66]、TASCC 结构形成[67]、促纤维化分子(如TGF-β 及FN)堆积[68]及激活纤维化通路[69]等起到促进肾纤维化作用。可见,细胞自噬对肾脏纤维化进程呈双向性影响,其原因可能包括:(1)细胞自噬在纤维化中的作用广泛;(2)自噬具有环境依赖性,在疾病的不同阶段可能发挥不同作用;(3)肾脏结构复杂,不同类型细胞对自噬激活反应不一致。因此,细胞自噬对AKI-CKD 转化及肾脏纤维化的作用与机制仍有较大的研究空间,并可能是AKI及AKI-CKD防治的新靶点[70]。
6.3 免疫相关性肾小球疾病 自噬参与免疫细胞的发育和分化并调节其功能[71]。目前研究表明,自噬在免疫相关性肾小球疾病进展中发挥重要作用。
6.3.1 IgA 肾病 在IgA 肾病患者肾活检组织中,可见足细胞内自噬体和肿胀溶酶体累积。在IgA 肾病小鼠模型中,SIRT1/SIRT3 介导的自噬活化可抑制IgA 免疫复合物刺激下巨噬细胞NLRP3 炎症小体的激活[72],提示免疫细胞自噬激活可能负向调控IgA肾病的发病。另外,系膜细胞作为IgA免疫复合物沉积后受到攻击的主要肾脏固有细胞,mTOR激活介导的自噬抑制可能是导致IgA 肾病系膜细胞增殖的主要原因[73],而采用mTOR 抑制剂激活细胞自噬,可减轻IgA 肾病大鼠系膜细胞损伤,提示系膜细胞自噬激活可减轻系膜细胞免疫炎症损伤。
6.3.2 膜性肾病 膜性肾病患者肾活检组织也可见足细胞自噬体大量堆积[58]。另外,膜性肾病患者可能由于血清中分泌型磷脂酶A2 IB 组(secretory phospholipase A2 group IB,sPLA2-IB)水平升高而激活足细胞M 型磷脂酶A2 受体(M-type phospholipase A2 receptor,PLA2R)所介导的p38 MAPK/mTOR/ULK1Ser757信号通路,导致足细胞自噬发生缺陷,最终引起肾病性蛋白尿[74]。
6.3.3 足细胞病 足细胞是免疫损伤重点打击的肾脏固有细胞之一,微小病变肾病和局灶节段性肾小球硬化症是两种经典的足细胞病。相比于局灶节段性肾小球硬化症患者的足细胞,微小病变肾病患者的足细胞显示出更高水平的自噬活性[75]。足细胞高水平的自噬活性往往预示着微小病变肾病病情稳定,不易于进展为局灶节段性肾小球硬化症。进一步的研究证实,沉积在肾小球上皮下的补体攻膜复合物C5b-9,可刺激足细胞,使其自噬溶酶体通路阻滞,这可能是膜性肾病患者蛋白尿发生以及持续的重要原因[76]。
6.3.4 狼疮性肾炎 在所有的免疫相关性肾小球疾病中,自噬与狼疮性肾炎的关系研究最多。Atg5的单核苷酸多态性影响狼疮的易感性[77]。在狼疮发生发展过程中,多种外周血循环免疫细胞自噬水平发生显著改变。研究发现:自噬调控狼疮自身反应性T、B 细胞的存活,也是浆母细胞分化所必需的;自噬参与介导狼疮患者Th17/Treg 的应答失衡[78];肾脏巨噬细胞自噬过度活化影响狼疮自身抗体产生和蛋白尿水平;自噬也可影响中性粒细胞外捕网(neutrophil extracellular traps,NETs)的形成和释放[79]。调控细胞自噬可再平衡免疫细胞的应答失衡,抑制炎症反应、减轻肾脏损伤。而且,狼疮状态下,足细胞自噬活性被激活,且足细胞自噬活性与足细胞损伤呈负相关[80]。应用雷帕霉素增加足细胞自噬活性,可减少狼疮自身抗体和干扰素α 诱导的足细胞凋亡及结构紊乱,说明自噬激活能对抗狼疮所致的足细胞损伤。
综上所述,目前自噬在免疫相关性肾小球疾病中的研究主要集中在两个层面:一是通过对免疫系统的影响而导致免疫相关性肾小球疾病的发生和发展;另一层是足细胞、内皮细胞和系膜细胞等自噬异常对疾病进展的影响。细胞自噬活性在免疫介导的肾小球不同疾病或同一疾病不同阶段的表现不同,目前也尚缺乏统一认识,需要进一步加强研究。
6.4 自噬与糖尿病肾脏疾病(diabetic kidney disease,DKD) DKD 发病机制涉及高血糖介导的血流动力学、细胞内代谢异常和细胞内应激等。其中氧化应激、内质网应激、营养过剩和缺氧等病理生理反应与细胞自噬异常关系密切[81]。足细胞在DKD 发病过程中起重要作用。DKD 早期,高糖可诱导足细胞自噬异常,抑制自噬会加速DKD 进展[54]。晚期糖基化终产物(advanced glycation end products,AGEs)抑制DKD状态下足细胞自噬活性:AGEs一方面通过损害溶酶体功能而抑制足细胞自噬,另一方面通过激活mTORC1 信号通路,抑制TFEB 活化,进而抑制足细胞的自噬活性,降低足细胞抗损伤能力[82]。此外,氧化应激等刺激因素也可通过抑制自噬体形成,抑制足细胞自噬活性,最终导致DKD 足细胞损伤和蛋白尿形成。另外,自噬在维持近端小管上皮细胞抵抗各种原因引起的细胞损伤中起重要作用。糖尿病患者肾活检组织中的小管上皮细胞及糖尿病小鼠小管上皮细胞均存在自噬受损,体外高糖刺激的近端小管上皮细胞自噬障碍[83]。进一步研究表明,高糖刺激的小管上皮细胞中mTOR 和Smad3 的过度激活分别抑制TFEB 活化及转录,造成溶酶体新生不足导致溶酶体耗竭,进而使自噬体无法通过溶酶体进行降解[38]。此外,DKD 所致的蛋白尿对肾小管上皮细胞的自噬也有影响,蛋白尿早期可促进肾小管上皮细胞自噬流,但持续大量的蛋白尿则可抑制肾小管上皮细胞自噬活性,此与损害自噬终末环节溶酶体的结构和功能相关[84]。值得注意的是,目前研究报道DKD 状态下肾脏细胞自噬反应水平不一,但一般认为在DKD 肾脏固有细胞中普遍存在自噬不足,其作用与机制仍有待进一步查明。

专家推荐意见六:1. 自噬在AKI、AKI-CKD 转化及肾脏纤维化过程中具有重要的调节作用,但其机制仍有较多的研究空间,建议进一步加强自噬在此领域的研究,并鉴定出新的调控分子及阐明其分子机制。2. 虽然有研究证明自噬通过对免疫细胞的影响在IgAN、膜性肾病、足细胞病、狼疮性肾炎等免疫相关性肾小球疾病进展中发挥重要作用,或通过对肾脏固有细胞自噬的影响而介导疾病发生发展,但目前尚缺乏统一认识,推荐进一步开展相关机制研究。3.DKD 不同时期肾脏固有细胞自噬反应水平不一,建议进一步加强自噬在DKD 不同时期、不同细胞类型中的精准作用与调节机制的研究。
7 靶向细胞自噬防治肾脏疾病
7.1 靶向mTOR 信号通路的药物 mTOR 是自噬启动阶段的关键调节因子,与多种肾脏疾病发生发展密切相关。雷帕霉素(rapamycin)是一种大环内酯类免疫抑制剂,同时也是最典型的mTOR 依赖性自噬诱导分子,因此,此药除了目前的用途之外,有望开发成为治疗以自噬活性抑制为特征的多种肾脏疾病的药物,事实上,多项研究证实雷帕霉素在顺铂诱导的AKI、糖尿病肾病和多囊性肾病等实验动物模型中具有肾脏保护作用[81],但需要进一步临床研究加以进一步验证。另外,作为mTORC1/mTORC2 双重抑制剂,Torin1 和PP242 调节自噬、保护肾脏损伤的作用也在动物实验中得以证实[85]。但需注意的是,mTOR对自噬和细胞生长分化有反向调节作用,一方面,mTOR 抑制剂可通过激活自噬保护肾脏,但也可能抑制细胞生长而阻碍肾脏损伤后的修复过程。另外,雷帕霉素过度诱导足细胞自噬可能与肾移植后患者出现蛋白尿有关,值得注意。
7.2 AMPK 激动剂 AMPK 激活是细胞自噬诱导的关键环节。二甲双胍可通过激活AMPK 而发挥自噬诱导作用、减轻AKI 时的肾小管上皮细胞损伤,且具有抗炎,抑制细胞凋亡、氧化应激、内质网应激和上皮-间充质转化的作用[86]。另外,二甲双胍可减轻糖尿病肾病患者足细胞丢失、系膜细胞凋亡和肾小管上皮细胞衰老,其机制也可能与调节自噬有关[87]。黄连素是一种异喹啉类生物碱,目前研究发现其可作为AMPK激活剂,通过诱导细胞自噬,抑制DKD肾小球系膜基质分泌及足细胞凋亡[88]。
7.3 靶向SIRTs SIRT1 是目前最受关注的SIRTs,也是近年来药物设计的新靶标。非黄酮类多酚化合物白藜芦醇作为SIRT1 经典激活剂,对AKI 及DKD具有肾脏保护作用[89]。SRT1720 是一种比白藜芦醇更有效的小分子SIRT1 激活剂,已被证实可改善顺铂诱导的AKI 及单侧输尿管梗阻引起的肾脏纤维化[90],对DKD 也具有一定的肾脏保护作用[91],其机制可能与调节细胞自噬有关,但仍需深入研究加以证实。
7.4 其它调节自噬的成分或药物 TFEB 是调控自噬活性、维持细胞自噬溶酶体通路稳定的关键转录因子,因此也是调节自噬的理想靶点。目前发现许多中药有效成分或提取物如姜黄素、木黄酮和海藻糖等具有激活TFEB 转录的作用。有报道姜黄素可通过抑制PI3K-AKT-mTOR 信号通路,激活TFEB 而增强自噬活性及溶酶体生物发生[92],且不依赖于mTOR 的抑制活性。木黄酮可激活TFEB 转录,增加其表达量,并通过抑制mTOR 活性而促进TFEB 磷酸化入核,通过改善溶酶体功能而增强自噬[93]。海藻糖通过抑制AKT 而激活AMPK,促使TFEB 活化入核,发挥调节自噬的作用。目前,上述药物通过调节自噬延缓肾脏疾病进展大多仍处于动物试验阶段,具有一定临床开发的前景。此外,锂和卡马西平等可降低细胞内磷酸肌醇水平,而米诺地尔和Tat-beclin 1 肽类药物可调节K+-ATP 通道开放[94]。它们已被证实在AKI、DKD、局灶性节段性肾小球硬化或多囊肾等动物模型中通过诱导肾脏固有细胞自噬而发挥肾脏保护作用[95]。
迄今为止,使用靶向细胞自噬研发的激动剂或抑制剂防治肾脏疾病仍然面临挑战,自噬在不同的肾脏疾病及不同的肾脏细胞类型,以及疾病进展不同阶段可能发挥着不同的作用,其详细调控机制和信号传递也有待进一步明确。目前自噬调节剂应用还缺乏临床试验证据,需要加快研究步伐。

专家推荐意见七:1. 靶向自噬通路有望成为肾脏疾病防治的新策略,建议进一步开展靶向mTOR、AMPK、SIRT 等信号通路的研究,探讨其对各种肾脏疾病的作用与机制。2. 推荐采用小分子肽类化合物或中药有效成分观察它们对自噬、自噬-溶酶体通路关键转录因子的调节作用及对实验性肾病的效果与机制。3. 建议在安全有效的前提下,逐步加强以自噬为靶点的肾脏病临床防治研究。
8 结语
随着对细胞自噬机制认识的不断深入以及检测手段的提升,细胞自噬在各种人类疾病中的作用越来越受重视,细胞自噬检测极有可能成为疾病诊断新手段,而自噬调控极有可能成为疾病防治的新措施。当前,自噬在肾脏疾病中的研究虽然也取得了一定成绩,但仍主要处于基础探索阶段,在肾脏疾病诊治中的应用还有待于开发。为此,我们制定了细胞自噬在肾脏疾病研究的理论和实践中国专家共识。希望本共识对有兴趣从事自噬与肾脏病研究的医务人员和科研人员在实际工作中有所帮助,并推动我国肾脏病领域自噬相关研究的发展,使自噬最终成为肾脏疾病诊治新策略。
附录
利益冲突:无。
专家组成员(按姓氏笔画排序):丁国华(武汉大学人民医院),王伟铭(上海交通大学附属瑞金医院),王惠明(武汉大学人民医院),刘友华(南方医科大学南方医院),刘华锋(广东医科大学附属医院),孙世仁(空军军医大学西京医院),庄守纲(上海同济大学附属东方医院),刘芳(四川大学华西医院),孙林(中南大学湘雅二医院),肖力(中南大学湘雅二医院),陈旻(北京大学第一医院),陈朝红(南京总医院),陆晨(新疆维吾尔自治区人民医院),杨俊伟(南京医科大学第二附属医院),张爱华(南京医科大学附属儿童医院),郑丰(大连医科大学附属第二医院),赵景宏(陆军军医大学第二附属医院),曾科(南京大学生命科学学院),焦军东(哈尔滨医科大学附属二院),谭小月(南开大学医学院),戴春笋(南京医科大学第二附属医院)
学术秘书:叶蓁楠(广东医科大学附属医院),周阳(南京医科大学第二附属医院)
猜你喜欢
中老年保健(2022年3期)2022-08-24
现代临床医学(2021年6期)2021-11-20
生物化工(2021年2期)2021-01-19
生物化工(2020年1期)2020-02-17
读与写(2019年35期)2019-11-05
中国生殖健康(2019年10期)2019-01-07
特别健康(2018年9期)2018-09-26
现代职业教育·高职高专(2018年7期)2018-05-14
中国病理生理杂志(2015年8期)2015-12-21
医学研究杂志(2015年3期)2015-06-10
