
曲妥珠单抗生物类似药临床有效性比对研究设计策略
2021-11-01 05:34关元宙王玉洁梁毅中国药科大学国际医药商学院南京211198
中南药学 2021年10期
关元宙,王玉洁,梁毅(中国药科大学国际医药商学院,南京 211198)
人表皮生长因子受体-2(human epidermal growth factor receptor-2,HER-2)是具有跨膜酪氨酸激酶受体的蛋白,其过量表达与肿瘤细胞的发生、进展及预后效果密切相关,曲妥珠单抗(trastuzumab)是一种人源化单克隆抗体,可与HER-2特异结合产生抗肿瘤作用[1]。该药商品名为赫赛汀,由Roche研制,最早于1998年获美国食品药品监督管理局(Food and Drug Administration,FDA)批准上市,目前在我国已获批3项针对HER-2阳性患者的适应证,包括早期乳腺癌(early breast cancer,eBC)、转移性乳腺癌(metastatic breast cancer,mBC)及转移性胃癌的治疗[2]。赫赛汀2019年全球销售额达60亿美金[3],其专利分别于2014年及2019年在欧洲及美国失效,巨大的经济利益吸引了约20家生物制药研发企业开展曲妥珠生物类似药的研发,其中不乏Amgen、Pfizer等跨国药企,还有Celltrion、复宏汉霖等新兴生物药企,目前已有8个曲妥珠单抗生物类似药在各地获批,仍有大批候选药处于药学及临床研究阶段。
鉴于曲妥珠单抗生物类似药临床研发日趋火热,本文整理了主要国家及地区生物类似药研发法规体系,同时归纳曲妥珠单抗生物类似药临床有效性比对试验的设计要点,以期为该类试验提供合理设计策略。
1 主要国家及地区生物类似药法规概况
2005年欧盟药品管理局(European Medicines Agency,EMA)通过了生物类似药上市许可申请指导原则,2009年美国通过的《生物制品价格竞争及创新法案》则加速了生物类似药研发浪潮。2015年国家药品监督管理局药品审评中心(Center for Drug Evaluation,CDE)发布了《生物类似药研发与评价技术指导原则(试行)》,为我国生物类似药研发提供技术指导及审评标准。各地法规及指导原则虽有细微差异,但大体上均覆盖生物类似药研发基本原则、药学研究、临床前研究、临床研究、上市后监测及适应证外推等内容。
2 文献检索及结果
在数据库“MEDLINE”“PubMed”“中国知网”及美国临床试验注册平台“ClinicalTrials”中以“曲妥珠”及“生物类似药”“trastuzumab”“biosimilar”等关键词进行检索,获得485篇文献,同时人工检索纳入其他3篇文献,通过去除重复及不相关文献,最终得到10篇已发表结果的曲妥珠单抗生物类似药临床有效性研究。本文按eBC新辅助治疗及mBC一线治疗两类适应证区分文献,归纳试验信息,结果见表1~2。
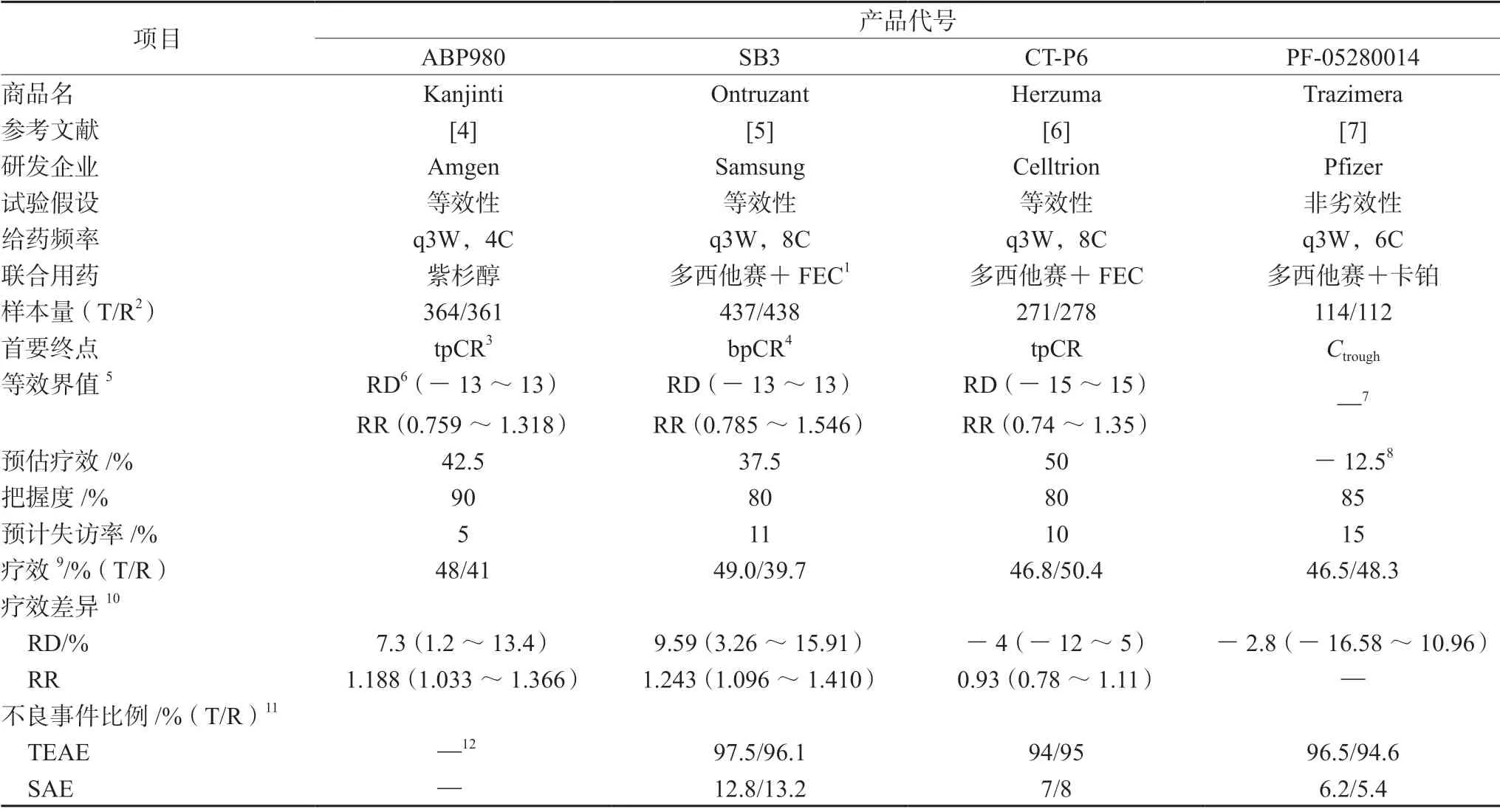
表1 以eBC新辅助治疗为研究人群及干预措施的临床研究信息汇总

表2 以mBC一线治疗为研究人群及干预措施的临床研究信息汇总
3 有效性比对试验设计要点
曲妥珠单抗生物类似药研发按照逐步递进原则,通过药学及临床前研究逐步确定候选药与赫赛汀可比后,再进行临床药理学研究验证体内代谢特征一致,最后开展临床比对试验证明两者有相似的有效性及安全性[14],本文从试验基本设计和统计分析两方面论述有效性比对试验设计。
3.1 试验基本设计
3.1.1 试验类型 EMA、FDA及CDE均推荐使用以原研药为对照的随机双盲等效性试验设计。等效性假设要求候选药有效性在预设区间内与赫赛汀相等,受等效上下限同时控制,足够的样本量能保证结论的可靠性。非劣效性区间仅为疗效下限,可证空间大,较小的样本量可能为结论带来偏倚,应慎重选用。
3.1.2 受试人群选择 应选择最敏感的人群进行研究,同时针对适应证及疾病背景进行纳排标准设计以保证研究人群的均质性。eBC新辅助治疗及mBC一线治疗的HER-2阳性患者为常用受试人群。eBC新辅助治疗患者前期未接受其他治疗,能敏锐检测出药物临床差异,已成为首选的试验人群[15]。mBC一线治疗患者是赫赛汀多项临床试验的研究人群,已累计大量临床数据可供使用,故其为常用受试人群,但应注意二线及以上治疗人群疾病背景复杂且对治疗不敏感,不利于比对临床有效性[16]。相对于乳腺癌治疗,曲妥珠单抗在转移性胃癌治疗中无明显临床产出,且中国胃癌患者HER-2阳性表达率低[17],应谨慎选择。
3.1.3 给药方案 mBC患者需联合紫杉醇或多西他赛等紫杉烷进行一线治疗,而eBC患者可用紫杉烷、FEC或卡铂等进行新辅助治疗,手术后继以辅助治疗。可采用3周一次(初始剂量8 mg·kg-1,后续剂量6 mg·kg-1)或每周一次(初始剂量4 mg·kg-1,后续剂量2 mg·kg-1)的给药频率,通常用药8周期。应在临床试验方案中明确患者缺失治疗后再次入组时治疗剂量及频率的调整方案,同时明确是否进行为期1年的维持治疗及适用人群。
3.1.4 有效性终点选择 抗肿瘤新药临床试验常采用无进展生存期(progression-free survival,PFS)、无病生存期(disease-free survival,DFS)及总生存期(overall survival,OS)等时间疗效终点来评价药物的有效性,但这类终点往往受患者疾病状况及过往用药等因素影响,无法满足灵敏度要求并使试验周期延长[18-19]。
EMA、FDA及CDE推荐使用ORR或病理完全缓解(pathological complete response,pCR)等能在短时间内体现药物临床差异的疗效指标作为首要终点[18-20]。ORR在晚期乳腺癌的临床治疗中与PFS展现密切相关性,是理想的代替性疗效指标,可在mBC一线治疗临床试验中代替PFS作为终点指标[21]。pCR能在较短的试验周期内准确预测eBC治疗效果,可作为eBC新辅助治疗临床试验药物加速审评临床试验的首要终点。FDA建议将pCR定义为乳腺及腋下淋巴结内恶性肿瘤和原位癌消失(tpCR),仅评价两部位恶性肿瘤的消失也可行,但需排除单纯的乳腺内恶性肿瘤消失(bpCR)[19]。无论选择何种人群及终点组合,都建议将PFS、DFS及OS列为次要终点供后期随访监测长期疗效[22]。
3.2 统计分析
3.2.1 有效性差异计算 体现药物临床有效性差异可使用RD或RR,两者作用相当,学术界及监管方面对采用哪一种表达形式未达成共识,如申报MYL-1401O时EMA要求使用RD体现,而FDA要求使用RR[10]。检索得到的10项试验中,有4项同时使用RD及RR两种形式以满足不同地区的监管要求,其他研究多以RD作为研究形式。相对不同的是,TrastuRel未使用RR或RD,仅通过对比双臂临床产出差异是否存在统计学意义作为判断两者临床等效的依据[13]。
3.2.2 等效界值设定 CDE指导原则[20]荟萃分析了3项赫赛汀联用紫杉烷对mBC一线治疗Ⅱ/Ⅲ期临床试验,得到两者ORR比值95%CI为1.92(1.544~2.386),保留50%疗效,最终确定候选药与赫赛汀RR 90%CI的等效界值为(0.8~1.25)。PF-05280014的mBC组荟萃了3项同样的临床研究,得到RR 95%CI等效界值为(0.80~1.25)[9]。而MYL-1401O虽然使用了相同临床数据,但疗效保留高于50%,故得出更严格等效值RR 90%CI(0.81~1.24)。尚无法规推荐eBC的等效界值,根据ABP980、SB3及CT-P6的eBC组的计算方法[4-6],可归纳出基本思路:文献荟萃估计赫赛汀联合用药较阳性对照的有效性,保留50%~60%的治疗效果以确定RD/RR的等效值。
目前各监管机构及业界尚未确定统一的等效值,且文献荟萃模型及计算假设均不一致,建议药企在进行临床试验设计时积极与监管机构进行沟通,避免造成结果误判。
3.2.3 样本量计算 根据荟萃分析预估曲妥珠单抗联合用药疗效,平衡试验假设、预估疗效、把握度及失访率计算样本量,按1∶1随机分配至候选药组及参照药组。建议荟萃分析纳入最新的高质量多中心临床试验,更准确预估不同适应证中曲妥珠单抗的疗效;CDE建议预设80%的把握度及10%的失访率[20]。10项临床试验样本量大小从几十至几百不等,归因于不同的试验假设和参数设定。等效性试验受等效上下限控制,要求大量样本,而非劣效性试验仅做单侧检验,所需样本较前者小;预估疗效、把握度及失访率设置越高,所需样本越大。PF-05280014的eBC组与TrastuRel选用了非劣效性假设,而TrastuRel及BCD-022仅参考一篇早期曲妥珠单抗临床试验[23]预估出过低的疗效,均导致总体样本量偏小。
3.2.4 统计分析口径 建议采用意向治疗集(intention-to-treat set,ITT),即满足纳排标准并随机入组的受试者,也采用随机入组后至少接受过1剂治疗的人群,ITT可能会稀释药物疗效但可以避免入组条件变更或病情恶化造成患者退出而带来的偏倚[24]。少数研究使用符合方案集(perprotocol set,PP),即在接受完整治疗方案的群体中进行有效性分析,在假设患者具备高度用药依从性的基础上充分反映药物间的疗效差异,剔除缺失治疗的患者可能导致有效性评估偏倚,应在临床试验方案中提前声明并合理解释原因,如SB3、CT-P6及PF-05280014的3项临床试验[5-7]。值得指出的是,ABP980药物试验选择随机入组后接受过治疗的人群作为分析样本,但由于各地人群基线存在差异,导致有效性结果溢出等效值,得出候选药疗效优于参照药的结论,后期通过敏感性分析平衡基线后,推断候选药与参照药等效[4]。
4 有效性比对试验设计策略
综合法规及过往临床试验可归纳出整体设计策略:在随机双盲等效性比对设计基础上,根据PICO框架从受试人群(population)、给药方案(intervention)、对照用药(control)、有效性终点(outcome)及纳排标准等方面确定曲妥珠单抗临床有效性比对试验方案,设计合理等效值及样本量,最后完成受试者入组并开展试验,详见图1。
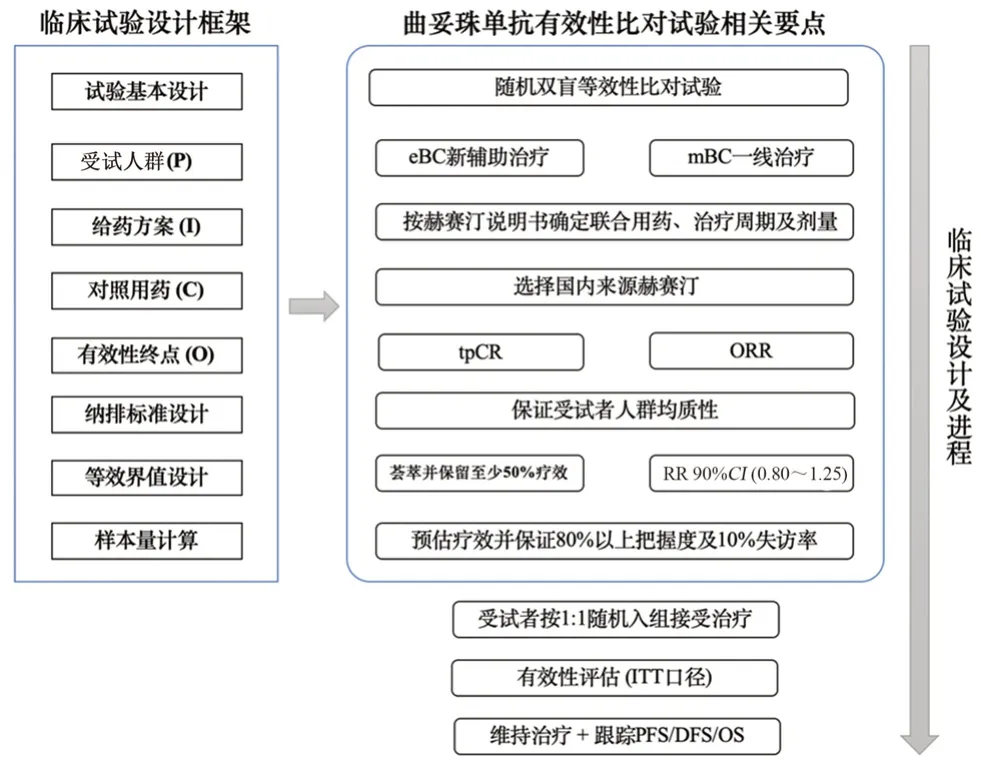
图1 曲妥珠单抗生物类似药有效性比对试验设计框架
5 结语
临床比对研究确立曲妥珠单抗生物类似药相似性后,可通过外推覆盖赫赛汀其余适应证[16],需要明确所选适应证与其余适应证具有相同病理机制及药物作用机制,同时评估候选药在新适应证上的安全性及免疫原性。曲妥珠单抗生物类似药上市后需根据赫赛汀不良反应进行用药安全监测,同时关注新不良反应信号,以确保用药安全。考虑到曲妥珠单抗需长期用药,建议研发企业对临床安全有效性进行延伸研究,探索其与赫赛汀的可互换性[25]。
我国是化学仿制药大国,发展生物类似药对研发企业的科研实力及药监部门的审评审批能力都提出了更高的要求。借助原研生物药专利到期的契机,我国药企投入研发,产出与原研药安全有效性相似的生物类似药,提高高值生物制品的可及性,带动新药研发,最终满足国民用药需求。
猜你喜欢
实用肿瘤学杂志(2022年3期)2022-11-30
基层中医药(2020年5期)2020-09-11
皮肤性病诊疗学杂志(2020年4期)2020-09-02
中国医药指南(2020年21期)2020-08-29
中外女性健康研究(2019年7期)2019-06-11
中外医疗(2017年27期)2017-11-15
中国老年保健医学(2016年6期)2017-01-09
中国卫生标准管理(2015年16期)2016-01-20
河南医学研究(2014年4期)2014-02-27
中国合理用药探索(2012年2期)2012-03-20
