
血管内皮细胞焦亡在动脉粥样硬化发生、发展中的作用及机制研究进展
2021-10-19 08:49金翠柳吴美平
医学研究杂志 2021年9期
柴 钰 金翠柳 凌 望 吴美平
动脉粥样硬化( atherosclerosis, AS)是一种以血管异常炎性反应为主要表现,以脂质沉积、斑块形成以及内膜增厚为主要病理特征,发生于血管内膜及内膜下的慢性炎症性疾病,是心绞痛、心肌梗死、心力衰竭、脑卒中等多种心脑血管疾病发生的主要病理基础[1]。内皮细胞是人体最大、最重要、功能高度活跃的器官,广泛分布于血管腔的内表面,其连续性和完整性是维持血管稳态的关键[2,3]。在动脉粥样硬化发生、发展的病理过程中通常伴有血管内皮细胞的死亡,研究表明,血管内皮细胞死亡是动脉粥样硬化形成的始动环节,同时存在于动脉粥样硬化发生、发展的整个病理进程[4]。
内皮细胞生存命运及其机制一直是冠心病病理学研究热点。迄今为止已发现的内皮细胞死亡方式包括凋亡、坏死、焦亡、铁死亡等,其中焦亡作为一种新的、独特的细胞死亡方式引起人们越来越多的关注。目前,在血管内皮细胞、血管平滑肌细胞、巨噬细胞和其他多种细胞类型中均发现细胞焦亡的存在,并且多项研究表明,细胞焦亡与动脉粥样硬化的形成密切相关。因此,深入探讨内皮细胞焦亡在动脉粥样硬化病理进程中的潜在作用和分子机制,可为动脉粥样硬化性心脑血管疾病的预防和治疗提供新的依据及策略。
一、血管内皮细胞之命运与动脉粥样硬化的关系
动脉粥样硬化是一种影响血管树并导致冠状动脉疾病(coronary artery disease,CAD)、外周动脉疾病和脑卒中的病理生理过程[5]。自1755年Albrecht von Haller提出“动脉粥样硬化”一词以来,人们对动脉粥样硬化病理生理学的理解有了很大的发展。现在认识到,最终导致冠心病的细胞和组织级联反应远比内皮细胞内层胆固醇颗粒的积累复杂得多。目前被普遍接受的由美国华盛顿大学医学院心血管病研究专家ROSS提出并修正的“损伤反应”学说认为,内皮细胞损伤是动脉粥样硬化早期病变发生的始动环节。
生理状态下,血管内皮细胞大多呈梭形,其排列与血流方向一致,可通过不同类型的黏附结构或连接方式形成连续的细胞单层,能有效隔离血液和组织液,使血管壁免受体内外各种物理、化学因素的损伤。但当血管内皮受到血流的冲刷、血脂紊乱、高血糖、烟草、药物、病原微生物等危险因素的刺激时,内皮细胞受损,内皮下空间中循环脂蛋白颗粒局部渗透、捕获和物理、化学修饰[6]。循环单核细胞从血液中选择性招募进入内膜,在内膜中它们分化成巨噬细胞并内化修饰过的脂蛋白成为泡沫细胞[7]。泡沫细胞进一步聚集形成脂纹,这是动脉粥样硬化的早期病变。动脉粥样硬化的病理变化过程可分为脂质条纹期、纤维样斑块期、粥样斑块期及复合病变期4个期,在一个特定的个体中,多个动脉粥样硬化斑块可以同时存在,每个斑块都处于其各自的病理生物进化阶段。在这一系列复杂事件中,内皮细胞扮演着极其重要的角色,内皮细胞功能减退乃至死亡构成了动脉粥样硬化致病的必要条件。
二、决定血管内皮细胞命运的方式
“生老病死”是生命的必经历程,亦是自然界的基本规律。但凡有生,终有归途。细胞死亡是细胞生命过程中的一个必经阶段,不仅对生物体的生长、发育具有重要的调节作用,而且对于维持生物体的稳态和基本生物学功能具有重要意义[8]。2009年细胞死亡学术命名委员会根据细胞形态学标准将细胞死亡分为坏死、凋亡、铁死亡、自噬及焦亡等[9]。
细胞坏死是一种裂解性细胞死亡,它由环境扰动触发,导致细胞核皱缩、质膜破裂、细胞自溶以及炎性细胞内容物失控释放[10]。细胞凋亡遵循细胞固有的程序性自杀机制,属于正常的细胞周期发育。其生理特征主要表现为细胞在不释放胞质内容物到细胞外空间的情况下自动解体并有效抑制炎症。铁死亡是一种氧化性非凋亡性的细胞死亡形式,其病理特征主要表现为细胞内铁依赖的脂质过氧化物蓄积,形态特征主要表现为细胞膜断裂出泡,线粒体体积变小,膜密度增加,嵴减少或消失,线粒体外膜破裂,但细胞核大小正常且无染色质凝聚[11]。自噬最早由著名细胞学家克里斯汀·德·迪夫提出,指膜包裹部分细胞质和细胞内衰老损伤的细胞器形成自噬体,并与溶酶体融合形成自噬溶酶体,降解其所包裹的内容物,以实现对细胞自身代谢需要的调控和细胞器的更新[12]。细胞焦亡(pyroptosis)是一种介于凋亡和坏死之间的炎症性程序性细胞死亡[13~15]。作为一种新进发现并被证实的细胞死亡模式,有研究证明,细胞焦亡依赖于半胱氨酸依赖性天冬氨酸特异性蛋白酶家族(caspase)而存在,且在形态、机制和病理生理上均不同于细胞凋亡和细胞坏死。焦亡发生时,孔道形成,质膜破裂,细胞内容物和促炎性介质进入细胞间质,激发机体的免疫反应,募集更多的炎性细胞进而扩大了炎性反应并导致细胞死亡。
三、细胞焦亡信号转导通路
1.经典细胞焦亡途径:经典细胞焦亡途径是依赖于caspase-1的细胞焦亡途径[16]。当细菌、真菌和病毒等病原体感染宿主细胞时,pro-caspase-1通过凋亡相关斑点样接头蛋白(apoptosis-associated speck-like protein containing CARD,ASC)与特异性模式识别受体(pattern recognition receptors,PRRs)NLR间接连接形成一个多聚体蛋白复合物炎性小体。pro-caspase-1一旦被募集到炎性小体后,它就会二聚体化并激活自身的蛋白酶活性,将无活性的caspase-1前体裂解形成有活性的caspase-1。活化的caspase-1一方面可以切割GSDMD使其成为GSDMD-N,后者能插入细胞膜诱导形成细胞膜孔道,这些孔道驱散细胞离子梯度,增加渗透压并导致渗透水流入,进而诱导发生细胞肿胀、质膜裂解、细胞死亡及胞内物质释放等一系列炎性反应。另一方面,活化的caspase-1也能切割白细胞介素-1β和白细胞介素-18前体,将其激活成为有活性的IL-1β和IL-18,并通过GSDMD细胞膜孔道将其释放至细胞外,招募更多炎性细胞,扩大炎性反应[17,18]。这种依赖caspase-1的细胞焦亡途径就被称为经典细胞焦亡途径。其中炎性小体的形成是经典细胞焦亡途径的重要特征之一。
2.非经典细胞焦亡途径:人的caspase-4/5和小鼠的caspase-11作为胞内受体,在受到革兰阴性细菌细胞壁脂多糖(lipopolysaccharide,LPS)刺激后,可识别LPS并与之直接结合而介导炎症坏死,这种依赖caspase-4/5/11的细胞死亡方式被称为非经典细胞焦亡途径[19]。其机制是LPS可通过与caspase-4/5/11结合而诱导其活化,活化的caspase-4/5/11在N末端和C末端结构域间的接头内直接剪切GSDMD,从而释放具有成孔活性的N端结构域GSDMD-N,GSDMD-N 通过结合膜磷酸肌醇转移到质膜,在膜上穿孔进而引起细胞焦亡[20]。此外,LPS与caspase-11特异性结合后,活化的caspase-11可促进缝隙连接半通道蛋白Pannexin-1的激活和Pannexin-1依赖性ATP通道的开放。裂解的Pannexin-1打开自身通道以释放ATP与细胞膜上P2X7 受体结合,打开非选择性P2X7阳离子通道,继而导致胞内的K+外流,胞外的Na+和Ca2+内流,细胞膜完整性受损,释放内容物引发炎性反应及细胞死亡[21]。虽然经典途径中的炎性小体也参与依赖caspase-11的细胞焦亡途径,但是caspase-11的自身活化与细胞死亡并不依赖于PRRs和ASC。因此,炎性小体并不是介导非经典细胞焦亡途径的必需条件。
四、血管内皮细胞焦亡与动脉粥样硬化
1.血管内皮细胞焦亡是促进动脉粥样硬化发生、发展的新机制:有研究表明,动脉粥样硬化病理变化过程中的多种体内外危险因素如高血压、高血糖、高血脂、吸烟及肥胖等均可导致内皮细胞焦亡。如Yin等[22]研究发现氧化低密度脂蛋白作用于内皮细胞后,可使内皮细胞中NLRP1、NLRP3、caspase-1和IL-1β表达水平均明显升高,诱导细胞焦亡。Xi等[23]研究发现内源性代谢刺激物同型半胱氨酸能够通过激活caspase-1依赖的炎性小体活化诱导细胞焦亡。另有研究报道,金属离子(Ni2+、Hg2+等)可导致内皮细胞NLRP3炎性小体活化、caspase-1激活和成熟的IL-1β产生,进而诱导细胞焦亡[24]。
内皮细胞焦亡可促进IL-1β、IL-18、P-选择素、细胞间黏附分子、血管细胞黏附分子等大量分泌,触发单核-吞噬细胞与血管内皮细胞的黏附,导致血管壁炎性反应的增强和血管内膜完整性的破坏,进而加剧动脉粥样硬化病变进程(图1)。如高胆固醇血症可通过NADPH氧化酶诱导ROS产生从而激活NLRP3炎性小体和caspase-1,诱导内皮细胞焦亡,促进炎性反应,加剧动脉粥样硬化[25]。吸烟是促使动脉粥样硬化发生的重要危险因素之一,Wu等[26]研究发现尼古丁可以促进ROS的产生,进而激活NLRP3炎性小体,促使caspase-1成熟和IL-1β、IL-18的产生,促进内皮细胞焦亡而加剧动脉粥样硬化。镉元素是一种较为常见的金属污染物,Chen等[27]研究发现,其可诱导线粒体产生ROS,进而活化NLRP3炎性小体及其下游的caspase-1和IL-1β,从而诱导人脐静脉内皮细胞焦亡,促进动脉粥样硬化的发生和发展。
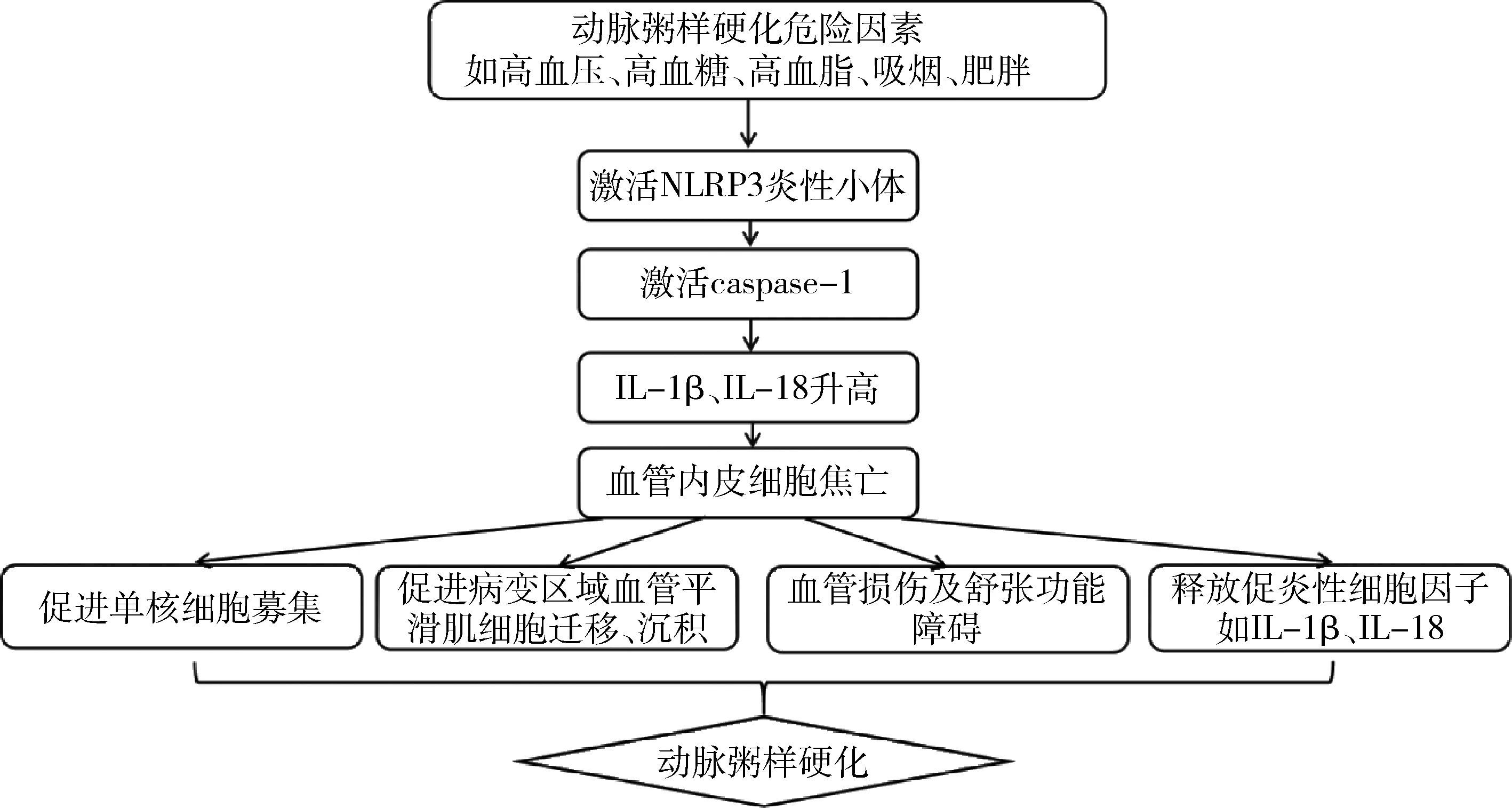
图1 血管内皮细胞焦亡与动脉粥样硬化
2.基于血管内皮细胞焦亡西医治疗动脉粥样硬化的现状:动脉粥样硬化是一种危及人类健康的慢性炎症性病变,是导致心脑血管疾病的主要原因。鉴于其临床与基础研究的不断深入,目前防治动脉粥样硬化的药物主要包括调脂类药物、抗栓和抗凝类药物、钙通道阻滞剂、抗氧化类药物等。调脂类药物如他汀类、贝特类、烟酸类、胆酸螯合剂等主要通过降低低密度脂蛋白发挥抗动脉粥样硬化的作用;抗栓类药物如阿司匹林、氯吡格雷、阿昔单抗等主要通过降低血小板的黏附和聚集防止血栓形成;抗凝类药物如普通肝素、低分子肝素、比伐卢定等通过影响凝血过程中的某些凝血因子抑制动脉粥样硬化斑块的形成;钙通道阻滞剂则主要通过减少低密度脂蛋白的氧化和平滑肌细胞的增殖,增加血管内皮舒张因子的释放,预防和控制动脉粥样硬化;抗氧化类药物如普罗布考和泛硫乙胺,可通过减少细胞因子和趋化因子对细胞的刺激作用,抑制细胞凋亡和坏死,从而阻断动脉粥样硬化的发生。
药物治疗的关键是发现靶点,通过靶点的抑制、激活和改变来达到阻止或逆转病程进展的目的。然而,虽然他汀类、贝特类和抗栓类等药物除靶向低密度脂蛋白和血小板外,还具有抗炎的功效,但对于血管内皮细胞焦亡这一新机制,目前尚无西药对其有深入明确的研究。
3.干预血管内皮细胞焦亡是中医药防治动脉粥样硬化的新策略:细胞焦亡是一种伴随炎性因子释放并引发细胞膜破裂溶解的新型死亡方式。近年来研究发现,该死亡形式在感染性疾病、代谢性疾病及动脉粥样硬化等多种疾病的发生、发展进程中扮演着重要角色。目前虽然用于干预细胞焦亡的药物种类不少,但多数还处于基础研究阶段,中医药作为经验医学,其研究与西医不同,多是在已有临床功能效果的基础下的机制研究。因此,中医药干预细胞焦亡的研究日益增多。洪蕾等[28]在探究很多中药如虎杖等的有效单体白藜芦醇对脂多糖诱导的人肺上皮细胞增殖、炎性因子释放及焦亡的影响时发现,白藜芦醇可以通过减少炎性因子的释放,降低NLRP3、Gasdermin D、caspase-1及ELAVL1的表达,对脂多糖诱导的人肺上皮细胞焦亡起到一定的保护作用。景艳芸等[29]研究发现,灯盏花乙素可通过上调巨噬细胞中cAMP水平来激活PKA,从而抑制ATP诱导的NLRP3炎性小体激活及巨噬细胞的焦亡。唐标等[30]以缺氧、缺糖(复氧)复糖诱导构建体外缺血再灌注SH-SY5Y细胞模型,探讨三七总皂苷对SH-SY5Y细胞焦亡的调控作用,结果发现三七总皂苷可降低细胞中GSDMD、GSDMD-N及caspase-1蛋白水平,抑制IL-1β和IL-18释放,进而减轻缺氧缺糖(复氧)复糖诱导的SH-SY5Y细胞功能损伤。中医药干预细胞焦亡或已成为新的研究热点。
血管内皮细胞作为血液与血管壁之间的保护性屏障,其焦亡容易导致血管内膜上脂质沉积,促使一系列的免疫反应导致白细胞沉积到血管壁,从而形成局部血栓。因此中医药针对血管内皮细胞焦亡的预防或治疗亦成为研究的热点之一。吴树宁等[31]在探索大黄素对脂多糖诱导的人脐静脉内皮细胞焦亡的影响时发现,大黄素可通过下调内皮细胞内活性氧水平来抑制NLRP3、caspase-1和IL-1β的表达,进而抑制缺氧条件下细胞焦亡,减少血管内皮损伤,防治心血管疾病。有研究采用二氢杨梅素预处理棕榈酸诱导的焦亡人脐静脉血管内皮细胞后,血管内皮细胞的活性及其细胞膜完整性增强,LDH释放减少,caspase-1、IL-1β和ICAM-1表达降低,内皮细胞焦亡减少。目前,细胞焦亡作为新的死亡方式已逐渐步入人们的视线,其释放的大量炎性物质及参与的多种蛋白成为可能的治疗靶点。中医药对血管内皮细胞焦亡的治疗与西医比较,对机体有更少的损害及不良反应。因此,中医药干预血管内皮细胞焦亡在防治动脉粥样硬化发生、发展中是一项新思路。
五、展 望
内皮细胞焦亡作为新进发现并被证实的一种伴随炎性因子释放的独特的程序性细胞死亡方式,其在内皮屏障和动脉粥样硬化发生、发展的病理进程中起着重要作用。在动脉粥样硬化病理进程中,多种体内外危险信号均可通过经典或非经典细胞焦亡途径引起血管内皮细胞发生焦亡,促进IL-1β、IL-18等炎性因子的大量释放,进而可以诱导炎性反应增强,促使斑块形成、易损和破裂。但由于研究处于开始阶段,使得目前研究相关性结论多,内皮焦亡与冠心病因果关系尚需开展深入研究予以证实。
中医对内皮细胞焦亡的研究目前处于基础阶段。因此,从中医药角度对内皮细胞焦亡的作用及机制进一步深入研究,发挥中医药的独特优势,通过中医药调控内皮细胞焦亡至阴阳平和状态同样必不可少。相信随着未来研究的不断深入,内皮细胞焦亡在动脉粥样硬化病理进程中的作用将逐步阐明,而内皮细胞焦亡也会成为解释中医药作用机制的有力证据。
猜你喜欢
中国医学工程(2022年8期)2022-11-25
中国神经免疫学和神经病学杂志(2022年1期)2022-11-25
重庆医学(2022年7期)2022-11-23
中西医结合心脑血管病杂志(2022年19期)2022-11-19
中国计划生育和妇产科(2022年6期)2022-11-15
中国临床解剖学杂志(2022年1期)2022-11-15
体育科技文献通报(2022年4期)2022-10-21
重庆医学(2022年14期)2022-08-06
检验医学与临床(2022年12期)2022-06-27
中国循证心血管医学杂志(2022年1期)2022-03-15
