
CDDO-Me对缺氧诱导的肺动脉外膜成纤维细胞活化的影响
2021-10-12 01:28彭丽瑶魏桂红黄张建解卫平
南京医科大学学报(自然科学版) 2021年9期
彭丽瑶,魏桂红,黄张建,孔 辉,解卫平*
1南京医科大学第一附属医院呼吸与危重症学科,江苏 南京 210029;2中国药科大学新药研究中心天然药物活性组分与药效国家重点实验室,江苏 南京 210009
肺动脉高压(pulmonary arterial hypertension,PAH)是一类以肺血管重构及肺血管阻力进行性升高为特征,右心室后负荷逐渐增加,最终导致右心衰竭甚至死亡的进展性疾病。肺血管重构是PAH发生发展的重要病理基础,主要表现为肺血管内膜增生、中膜肥厚、外膜纤维化及炎症细胞浸润等[1]。研究表明,肺动脉外膜是最早响应血管应激或损伤并发生病理改变的结构[2]。在缺氧、血管扩张等各种因素的作用下,肺动脉外膜成纤维细胞(pulmonary artery adventitia fibroblast,PAAF)作为肺动脉外膜中主要的细胞成分首先被激活,主要表现为细胞的增殖、迁移能力显著增强,并可以向肌成纤维细胞转化,产生更多的细胞外基质(extracellular matrix,ECM)、生长因子、趋化因子以及炎性细胞因子,调节血管壁细胞的生长,参与肺血管重构过程[3]。
缺氧可以诱导细胞产生大量活性氧(reactive oxygen species,ROS),当ROS 的产生超过了机体抗氧化防御能力时,组织细胞便会出现基因突变、凋亡坏死、脂质过氧化等一系列氧化应激损伤。研究表明,氧化应激可以诱导肺血管过度收缩、肺动脉内皮功能障碍以及肺血管重构,参与PAH的发生发展[4]。核转录相关因子-2(nuclear factor erythroid-2 related factor 2,Nrf2)是细胞防御氧化应激的主要转录调节因子,在维持细胞的氧化还原平衡及代谢中发挥重要作用[5]。甲基巴多索隆(bardoxolone methyl,CDDO-Me)是一种新型三萜类齐墩果酸衍生物,可激活Keap1/Nrf2/ARE 信号通路发挥抗氧化应激作用,在慢性肾病[6]、2型糖尿病[7]、急性肺损伤及肺纤维化[8-9]中具有潜在治疗作用。然而目前尚无研究报道其对缺氧诱导的PAAF的影响。本文旨在探索CDDO-Me 对缺氧诱导的PAAF 活化后功能的影响并探讨其相关机制。
1 材料和方法
1.1 材料
人PAAF、成纤维细胞培养基(ScienCell 公司,美国),CDDO-Me(中国药科大学黄张建教授惠赠),CCK-8 试剂盒(同仁公司,日本),转化生长因子(transforming growth factor,TGF)-β1、肿瘤坏死因子(tumor necrosis factor,TNF)-α、白细胞介素(interleukin,IL)-1β ELISA 检测试剂盒(武汉华美生物),2′,7′-二氯荧光黄双乙酸盐(DCFH-DA,Sigma公司,美国),总谷胱甘肽(glutathione,GSH)检测试剂盒、丙二醛(malondialdehyde,MDA)检测试剂盒(上海碧云天生物),总超氧化物歧化酶(superoxide dismutase,SOD)测定试剂盒(南京建成生物工程研究所)。βactin 抗体、α-平滑肌肌动蛋白(alpha-smooth muscle actin,α-SMA)抗体、Ⅰ胶原蛋白(Collagen Ⅰ)抗体、羊抗兔/羊抗鼠二抗(武汉Proteintech 公司),波形蛋白(Vimentin)抗体(Abcam 公司,美国),NF-κB pp65/p65 抗体、p-Smad3/Smad3 抗体(CST 公司,美国),羊抗兔荧光二抗(Invitrogen公司,美国)。
1.2 方法
1.2.1 细胞培养及分组
人PAAF 培养于成纤维细胞完全培养基中(含2%胎牛血清、1%成纤维细胞生长因子、100 U/mL青霉素及链霉素),置于37 ℃、5%CO2的培养箱中培养。细胞生长至70%融合时分组处理,分为4组:常氧组、缺氧组、缺氧+CDDO-Me 组、常氧+CDDO-Me组,其中缺氧组和缺氧+CDDO-Me组置于含5%CO2、1%O2缺氧箱中处理。CDDO-Me在放入缺氧箱前1 h加入。
1.2.2 CCK-8法检测细胞活力
收集对数生长期的人PAAF,按每孔1×104个细胞接种于96 孔板,常规培养24 h 后弃培养基,按照分组分别加入无血清培养基和无血清培养基配制的不同浓度CDDO-Me工作液。1 h后,将缺氧组及缺氧+CDDO-Me组置入缺氧箱中培养。24 h后,弃培养基,每孔加入含10%CCK-8原液的培养基100 μL,孵育4 h,应用酶标仪于450 nm 波长处检测吸光度。细胞活性=(实验组吸光度值-空白组吸光度值)/(对照组吸光度值-空白组吸光度值)×100%。
1.2.3 Transwell迁移实验
收集对数生长期的人PAAF,PBS洗涤3遍后用无血清培养基将其重悬制成1×105个/mL 的细胞悬液,在Transwell 上室中加入100 μL 细胞悬液,下室中加入600 μL 含所需药物的完全培养基。根据分组,置于不同培养箱培养。24 h后取出小室,弃去上室培养基,用棉签擦去小室膜上层细胞,多聚甲醛固定,PBS 洗涤,结晶紫染色后于显微镜下观察,使用Image-Pro Plus 图像分析软件分析每组迁移至下室的细胞所占面积,计算细胞相对迁移率。
1.2.4 细胞免疫荧光分析
人PAAF以5×104个/孔的密度接种于24孔板细胞爬片上,隔天弃去培养基,药物处理1 h后分别置于不同培养箱中培养24 h,PBS清洗后多聚甲醛固定30 min,0.5%TritonX-100 PBS液洗涤20 min,5%BSA室温封闭1 h,随后加入一抗α-SMA、p65(1∶200),置于4 ℃冰箱孵育过夜,PBS清洗3次后加入免疫荧光二抗(1∶1 000),置于室温孵育1 h。PBS清洗后每孔加入Hoechst 33342染核30 min,PBS清洗后用抗荧光淬灭液封片,在荧光显微镜下观察拍照。
1.2.5 Western blot
收集处理后的人PAAF,PBS洗3遍后加入预冷的RIPA裂解液提取细胞总蛋白。常规SDS-PAGE凝胶电泳、湿法转膜,5%脱脂奶粉溶液室温封闭1 h,随后加入一抗(1∶1 000)置于4 ℃冰箱孵育过夜。TBST 洗膜后加入二抗(1∶8 000),室温孵育1 h,洗膜后化学发光显影,使用Image Lab图像分析软件分析蛋白表达量。
1.2.6 氧化应激水平检测
ROS 检测:按照1.2.4 方法培养细胞,24 h 后弃去培养基,每孔加入无血清培养基稀释的DCFH-DA(10 μmol/L),37 ℃细胞培养箱内孵育20 min。用无血清培养基洗涤细胞3 次,置于荧光显微镜下观察拍照。
GSH、MDA 和SOD 检测:按照1.2.5 方法收集细胞裂解液,按照试剂盒说明书操作,GSH 检测在412 nm 波长处测定吸光度、MDA 检测在532 nm 波长处测定吸光度、SOD检测在450 nm波长处测定吸光度。
1.2.7 酶联免疫吸附实验(ELISA)
人PAAF 以5×104个/孔的密度接种于24 孔板中,隔天弃去培养基,药物处理1 h后分别置入不同培养箱培养,药物处理24 h后收集细胞上清,-80 ℃保存备用。取出细胞上清和ELISA检测试剂盒室温复温30 min,按照试剂盒说明书操作,应用酶标仪于450 nm 波长处检测各孔吸光度值。应用CurveExpert 软件制作标准曲线,根据样本吸光度值计算细胞因子浓度。
1.3 统计学方法
采用SPSS 22.0统计软件进行分析,所有数据以均数±标准误()表示,多组间样本统计分析采用单因素方差分析(one-way ANOVA),组间两两比较采用LSD法。P<0.05为差异有统计学意义。
2 结果
2.1 CDDO-Me 对缺氧诱导的PAAF 细胞活力和迁移的影响
CCK-8实验结果显示,在常氧条件下,500.0 nmol/L及以下浓度的CDDO-Me 对细胞活力的影响与对照组相比差异无统计学意义(P>0.05,图1A),表明500.0 nmol/L 及以下浓度的CDDO-Me 对PAAF 无细胞毒性。缺氧刺激24 h 可以显著增强PAAF 细胞活力,与常氧组相比差异有统计学意义(P<0.05,图1B);CDDO-Me 可以浓度依赖性地降低缺氧诱导的PAAF 细胞活力的升高,并在250.0 nmol/L 和500.0 nmol/L 浓度时具有显著抑制作用(P<0.05),故使用500.0 nmol/L 的CDDO-Me 进行后续研究。Transwell 迁移实验结果显示,与常氧组相比,缺氧组细胞迁移数量显著增加,而缺氧+CDDO-Me 组细胞迁移数量显著减少(P<0.05,图1C、D),提示CDDO-Me 可以显著抑制缺氧诱导的PAAF 迁移。上述结果表明,缺氧可以诱导PAAF 细胞活力和迁移能力增强,而CDDO-Me可以显著抑制上述改变。
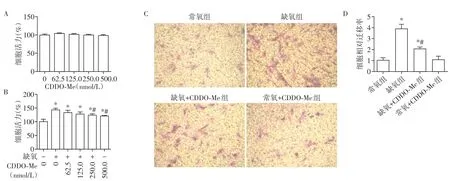
图1 CDDO-Me对缺氧诱导的PAAF细胞活力和迁移的影响Figure 1 Effects of CDDO-Me on the viability and migration of PAAF induced by hypoxia
2.2 CDDO-Me 对缺氧诱导的PAAF 向肌成纤维细胞转化的影响
细胞形态学结果显示(图2A),在常氧条件下PAAF呈现细长纺锤状,细胞整体长度较长,宽度较窄。在缺氧刺激24 h后,PAAF的胞体宽度增加,细胞呈现肥大改变,而缺氧+CDDO-Me 组的PAAF 细胞肥大程度得到改善。同时,细胞免疫荧光结果显示(图2B),与常氧组相比,缺氧组PAAF内α-SMA荧光强度增强,而缺氧+CDDO-Me 组α-SMA 荧光强度相较于缺氧组显著减弱。Western blot 半定量结果进一步表明,缺氧可以显著上调PAAF 中肌成纤维细胞标志物Collagen Ⅰ、Vimentin 和α-SMA 的表达水平,与常氧组相比差异有统计学意义(P<0.05,图2C~E)。而缺氧+CDDO-Me 组PAAF 的Collagen Ⅰ、Vimentin和α-SMA的表达水平与缺氧组相比显著下降(P<0.05)。以上结果表明,CDDO-Me 可以抑制缺氧诱导的PAAF向肌成纤维细胞转化。
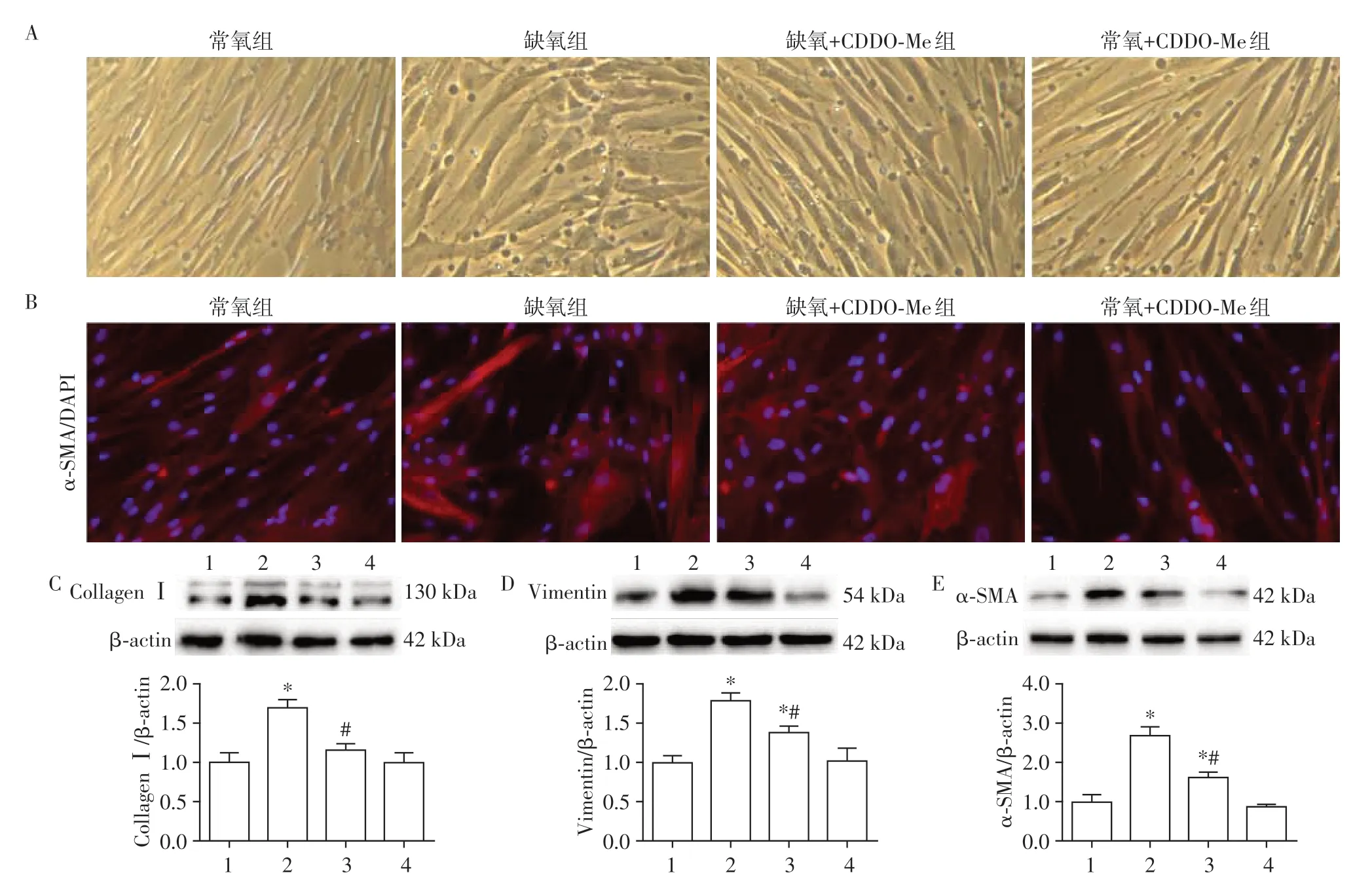
图2 CDDO-Me对缺氧诱导的PAAF向肌成纤维细胞转化的影响Figure 2 Effects of CDDO-Me on the myofibroblast transformation of PAAF induced by hypoxia
2.3 CDDO-Me对缺氧诱导的PAAF氧化应激的影响
ROS 检测结果显示,与对照组相比,缺氧组PAAF 中ROS 荧光强度显著增强(P<0.05,图3A、B),提示缺氧显著上调细胞内ROS 水平,而CDDOMe可显著抑制缺氧诱导的ROS升高,与缺氧组相比差异有统计学意义(P<0.05)。此外,缺氧刺激24 h后,PAAF 中反映细胞氧化损伤程度的重要指标MDA 的表达水平显著上调,发挥细胞抗氧化作用的GSH和SOD表达水平显著下调(P<0.05,图3C~E)。与缺氧组相比,CDDO-Me可以显著抑制缺氧诱导的MDA产生,并上调GSH和SOD表达水平(P<0.05)。
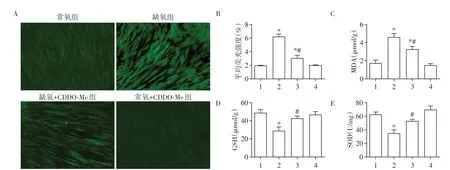
图3 CDDO-Me对缺氧诱导的PAAF氧化应激的影响Figure 3 Effects of CDDO-Me on the oxidative stress of PAAF induced by hypoxia
2.4 CDDO-Me 对缺氧诱导的PAAF 中TGF-β1/Smad3信号通路的影响
ELISA(图4A)及Western blot 结果(图4B)显示,与对照组相比,缺氧组PAAF合成分泌的细胞因子TGF-β1水平显著升高,Smad3磷酸化水平显著上调(P<0.05)。与缺氧组相比,缺氧+CDDO-Me 组PAAF 的TGF-β1 表达水平以及Smad3 磷酸化水平显著下降(P<0.05)。以上结果提示,CDDO-Me 可以抑制缺氧诱导的PAAF 中TGF-β1/Smad3 信号通路的激活。
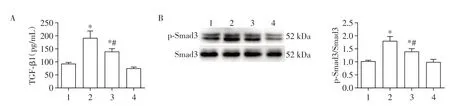
图4 CDDO-Me对缺氧诱导的PAAF中TGF-β1/Smad3信号通路的影响Figure 4 Effects of CDDO-Me on the TGF-β1/Smad3 signaling pathway in PAAF induced by hypoxia
2.5 CDDO-Me 对缺氧诱导的PAAF 中NF-κB 信号通路的影响
免疫荧光结果显示(图5A),常氧组PAAF中NFκB(p65)主要表达于细胞质中,缺氧组NF-κB 主要表达于细胞核中,表明缺氧可诱导NF-κB向细胞核转位,而CDDO-Me 处理后可抑制上述改变。Western blot和ELISA结果进一步表明,与对照组相比,缺氧组PAAF 中NF-κB 磷酸化蛋白水平显著上调,同时NF-κB 信号通路下游靶基因IL-1β和TNF-α的表达水平显著升高(P<0.05,图5B~D)。与缺氧组相比,CDDO-Me处理可以显著降低NF-κB磷酸化水平,并下调IL-1β和TNF-α的表达水平(P<0.05)。以上结果表明,CDDO-Me可以抑制缺氧诱导的PAAF中NF-κB信号通路的激活。
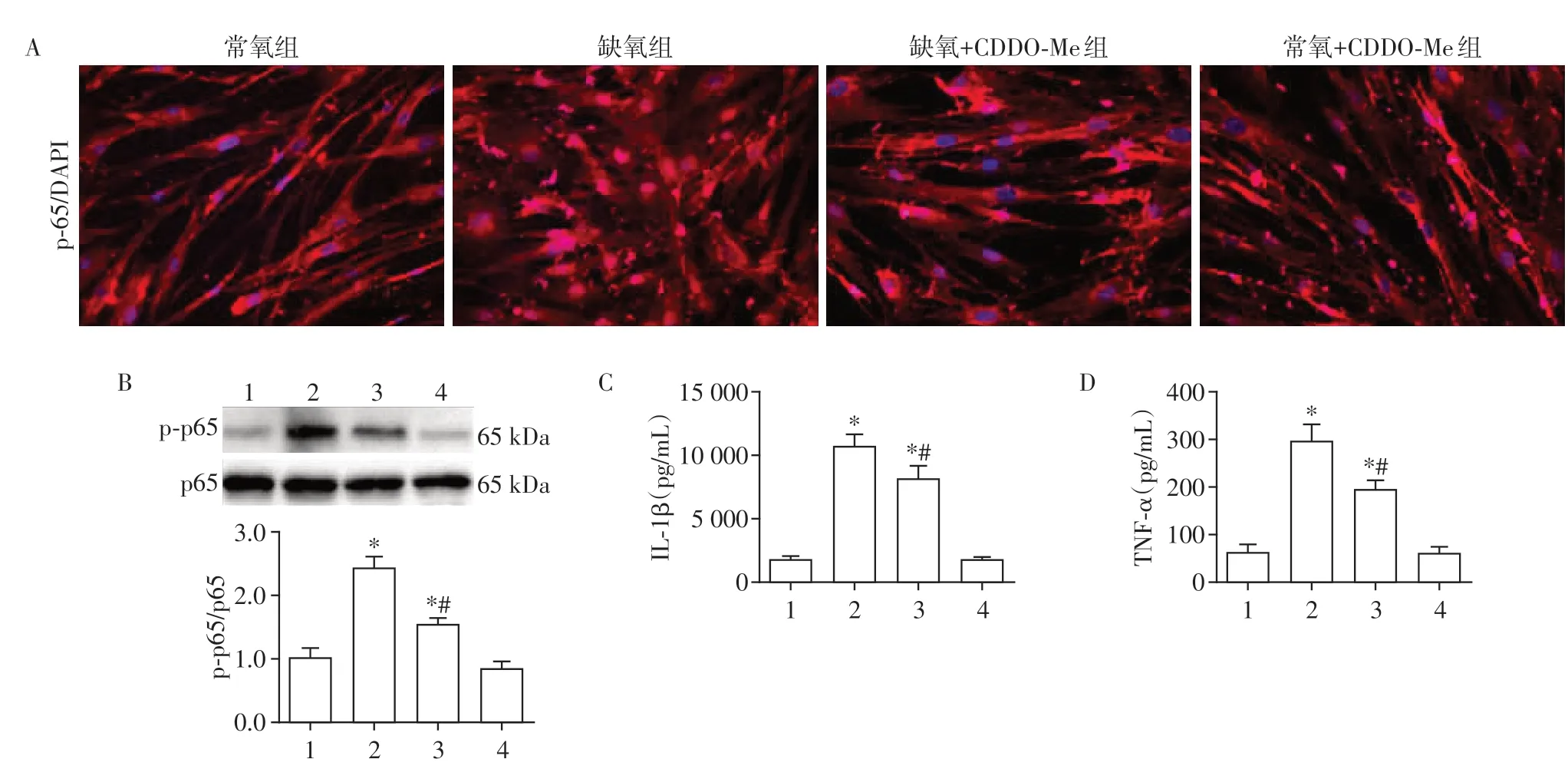
图5 CDDO-Me对缺氧诱导的PAAF中NF-κB信号通路的影响Figure 5 Effects of CDDO-Me on the NF-κB signaling pathway in PAAF induced by hypoxia
3 讨论
肺血管重构是PAH特征性的病理改变,涉及肺血管内膜和中膜增厚以及血管外膜的纤维化。肺动脉内皮细胞功能障碍和平滑肌细胞过度增殖是PAH的研究重点,而成纤维细胞作为血管外膜最丰富的细胞成分,其在PAH发生发展中的作用常被忽略。近年来越来越多的研究表明,在响应损伤和应激时,血管外膜成纤维细胞最先被激活,在肺循环和体循环血管结构的调节中发挥重要作用[2,10]。尤其是在缺氧条件下,肺血管外膜常在肺动脉中膜增厚以及肺血管压力升高之前便出现结构改变,包括成纤维细胞的过度增殖与迁移、肌成纤维细胞转化、大量胶原蛋白的沉积以及ECM的重构[11]。这种改变不仅使血管壁顺应性降低,同时ECM作为多种大分子蛋白组成的复杂网状结构,其成分变化参与调节血管壁细胞增殖、迁移、分化等多种生物学过程[12]。
氧化应激损伤在缺氧性PAH 中发挥至关重要的作用。缺氧刺激血管壁细胞产生大量ROS,同时细胞内清除ROS的能力下降,细胞自身的氧化/抗氧化平衡失调造成大量ROS蓄积,诱导细胞发生炎症、凋亡、坏死,促进血管重构[13]。ROS还可以与一氧化氮(nitric oxide,NO)相互作用降低NO的生物利用度,增加血管张力[14]。Nrf2是细胞抗氧化应激的重要转录因子,调控下游如GSH、硫氧还蛋白、醌氧化还原酶等抗氧化相关蛋白及细胞保护酶的表达,在维持细胞氧化还原稳态中发挥重要作用。CDDO-Me 是一种半合成的齐墩果酸衍生物,可以有效激活Keap1/Nrf2/ARE信号通路,诱导抗氧化相关蛋白及酶类的表达,降低ROS水平,保护细胞功能[15]。研究显示,CDDO-Me 可以减轻缺血再灌注引起的大鼠急性肾损伤[16]、保护糖尿病肾病肾小管功能[17],也可以减轻放射线照射和博来霉素诱导的小鼠肺部损伤和纤维化[8-9]。此外近年来研究表明,CDDO-Me 可改善内皮细胞功能、抑制平滑肌细胞收缩[18]。Ⅱ期临床试验结果显示CDDO-Me可改善PAH患者运动耐力[19]。以上研究均提示CDDO-Me 具有较大的PAH治疗潜力。本课题组前期研究结果表明,CDDO-Me可降低野百合碱诱导的PAH大鼠平均肺动脉压,在PAH 动物模型中具有一定治疗作用[20]。然而目前CDDO-Me对PAAF生物学功能的影响尚不明确。
肺血管外膜纤维化是肺血管重构的一个重要病理改变。在缺氧性PAH早期,肺动脉外膜便出现大量表达α-SMA 的肌成纤维细胞。肌成纤维细胞作为胶原蛋白和其他ECM蛋白的主要产生细胞,在诱导血管外膜纤维化、降低血管顺应性中发挥重要作用[21]。此外,肌成纤维细胞具有更强的收缩和迁移能力,参与血管新生内膜的形成[22]。本研究结果显示,CDDO-Me 可以抑制缺氧诱导的PAAF过度增殖、迁移和肥大,抑制α-SMA、Vimentin和Collagen Ⅰ的表达,提示CDDO-Me 能够抑制缺氧诱导的PAAF向肌成纤维细胞转化。
成纤维细胞向肌成纤维细胞的转化过程受到复杂的微环境调控,包括生长因子、细胞因子、黏附分子和ECM 分子。研究表明,缺氧可以诱导NADPH氧化酶(NADPH oxidases,Nox)表达增加,进而产生大量ROS,促进成纤维细胞向肌成纤维细胞转化[23]。抑制Nox4 或应用抗氧化剂N-乙酰半胱氨酸降低细胞内ROS 水平可以改善博来霉素诱导的肺纤维化[24]。本研究结果表明,CDDO-Me可以显著升高细胞中GSH水平,增加抗氧化金属酶SOD的表达,减少缺氧诱导的ROS 蓄积和脂质过氧化产物MDA 的生成,提示CDDO-Me 可以提高细胞抗氧化应激能力。此外,缺氧还可以通过上调缺氧诱导因子-1α(hypoxia inducible factor-1α,HIF-1α)的表达来促进TGF-β1 的分泌,促进纤维化进程[25]。TGF-β1是强有力的促纤维化细胞因子,其下游底物分子Smad3 可以结合促纤维化相关蛋白的启动子区域,促进α-SMA和胶原蛋白的表达,诱导基质金属蛋白酶组织抑制因子的产生并抑制基质金属蛋白酶的活性,从而减少ECM降解,因此TGF-β1/Smad3信号通路的过度活化是纤维化疾病发生发展的重要机制之一[26]。本研究结果证实,缺氧可以促进PAAF分泌细胞因子TGF-β1,激活下游Smad3 信号通路,而CDDO-Me能够抑制缺氧诱导的TGF-β1/Smad3信号通路的活化。以上实验结果表明,CDDO-Me可能通过减少PAAF 中ROS 的蓄积,抑制缺氧诱导的TGF-β1/Smad3 信号通路的活化来调控PAAF 向肌成纤维细胞转化。
此外,成纤维细胞可以分泌多种炎性因子[27],研究表明,其可能通过激活STAT3、C/EBPβ等信号通路,诱导巨噬细胞的招募与激活,促进巨噬细胞向炎症表型转化,参与PAH 的发生发展[28]。NF-κB活化是炎症反应的中心环节,静息状态下NF-κB与IκB 结合存在于胞浆中,其功能受到抑制。当细胞外信号刺激时,IκB被泛素化降解,NF-κB得以解离并转位至细胞核中,调节炎症趋化因子如单核细胞趋化蛋白-1和多种炎症细胞因子(如IL-6和TNF-α)的表达,在炎症、氧化应激、细胞增殖与凋亡等过程中发挥重要作用[29]。本研究结果显示,缺氧可以激活PAAF中NF-κB信号通路,诱导NF-κB的核转位,升高NF-κB 蛋白磷酸化水平并促进炎症因子IL-1β和TNF-α的合成释放。而CDDO-Me 可以抑制缺氧诱导的PAAF 中NF-κB 信号通路的活化,降低下游炎症因子IL-1β和TNF-α的表达。这一结果与既往研究相一致,即Nrf2信号通路与NF-κB信号通路之间具有相互作用,Nrf2信号通路的激活可以抑制NFκB的活化以及相关炎症因子的表达[30],同时CDDOMe也在多种疾病模型中表现出抗炎作用[15]。
综上所述,CDDO-Me 可以抑制缺氧诱导的PAAF增殖、迁移以及肌成纤维细胞转化,其可能与提高PAAF 抗氧化应激能力、降低ROS 水平并抑制TGF-β1/Smad3信号通路相关。CDDO-Me 还可以抑制缺氧诱导的NF-κB 信号通路活化,减少PAAF 炎症因子的释放,发挥抗炎作用。现有的PAH治疗药物在改善肺血管重构方面作用有限,以CDDO-Me为载体研发的CDDO-NO,兼具扩张肺血管和抑制肺血管重构的作用,在野百合碱诱导的PAH大鼠模型中具有一定治疗效果[20]。在今后的研究中,仍需进一步探索CDDO-Me 及以其为载体的生物合成药物更深层次的分子作用机制以及信号通路,为PAH的治疗提供新的方向。
猜你喜欢
昆明医科大学学报(2022年4期)2022-05-23
上海交通大学学报(医学版)(2022年3期)2022-05-05
井冈山大学学报(自然科学版)(2022年1期)2022-02-28
商品与质量(2021年27期)2021-11-23
昆明医科大学学报(2021年8期)2021-08-13
云南医药(2021年3期)2021-07-21
药学研究(2021年3期)2021-04-20
心肺血管病杂志(2020年5期)2021-01-14
食品安全导刊(2020年21期)2020-12-03
食品安全导刊(2020年18期)2020-12-03
