
拟南芥XTH33 基因的CRISPR/Cas9高效敲除质粒构建
2021-10-11 08:36王大鹏孔丽娜闫金国
花卉 2021年20期
王大鹏,孔丽娜,黄 姗,闫金国
(潍坊护理职业学院医学基础部,山东 青州 262500)
近年来的研究成果深化了我们对于木葡聚糖生物合成、重排及降解的酶学认识。研究发现木葡聚糖内糖基转移酶/水解酶(xyloglucan endotransglucosylase/hydrolase, XTH)为多基因家族编码的一类蛋白质,属于糖苷水解酶家族GH16。其对植物根茎叶的形态建成、木质部的形成中发挥重要作用,并能影响植株果实发育,增强植株抗逆性。
人工改造过的Cas9/gRNA 系统通过gRNA 引导Cas9 蛋白识别并剪切带有gRNA 靶点的双链DNA,用于基因敲除和精确编辑DNA 等操作。CRISPR 技术进行基因敲除和编辑操作的第一步,需要获得剪切活性高的gRNA 靶点。Cas9/gRNA 剪切活性是由gRNA 靶点的识别序列决定的,每个gRNA 的剪切活性都会不同。本研究经体外gRNA 活性检测,选取高效靶点,成功构建拟南芥XTH33 基因敲除质粒,并将敲除质粒转入农杆菌GV3101,为进一步进行XTH33 基因功能鉴定奠定基础。
1 材料与方法
1.1 材料
gRNA 靶点效率检测试剂和pUbi-atU6-gRNA 质粒骨架均由北京唯尚立德生物科技有限公司购买,DH5α 感受态细胞、Loading Buffer、DNAmarker、T4 磷酸化酶(T4PNK)、质粒纯化试剂盒、PBS 缓冲液、成品LB 培养基粉末、琼脂糖粉、农杆菌GV3101、T4连接酶、引物合成及测序由博迈德生物科技有限公司完成。
1.2 XTH33 基因的gRNA 设计与合成
首先应用在线工具(http://crispr.mit.edu)设计出符合实验要求的gRNA,gRNA 设计的主要原则[2]:必须选择在靠前的外显子区域;3’端要20 个连续的碱基序列,有NGG 碱基序列但不包括PAM 序列;并用生物信息学软件对拟南芥进行全基因组比对,尽量降低脱靶风险,以防Cas9 无法工作,需避免染色体和核小体三维结构造成的空间位阻效应,设计完成后合成引物gRNA-G1-FP1、gRNA-G2-FP2、gRNA-G3-FP3、gRNA-RP。
1.3 体外gRNA 靶点活性检测
四 条 引 物gRNA-G1-FPg1、gRNA-G2-FPg2、gRNA-G3-FPg3、gRNA-RP,分别使用T7-gRNA-FPg(1-3)和gRNA-RP 引物对,以标准gRNA 模板为模板做PCR,对于每个样品50μL 体系扩增2 管,检测正确后使用PCR 产物纯化试剂盒进行过柱纯化操作,最后使用40μ LDEPC H2O 洗脱DNA,测浓度(至少要≥70ng/μL)作为后续体外转录的DNA 模板。同时准备标准gRNA1(g1)和标准gRNA2(g2)的PCR 产物。酶切DNA 的准备:将gRNA 靶点(包括PAM 序列)通过搭桥PCR 的方式构建到PCR 产物中,回收备用。gRNA 的转录:10×Transcription Buffer 2μL、rNTPT7 2μL、RNA Polymerase Mix 2μL、gRNA PCR 产物(上一步产物)1μg、DEPC H2O up to 20μL,以上混合后,37℃反应2H,反应结束后,加入2μL DNase I,37℃反应30min,并对gRNA 进行回 收 与 提 取。Cas9/gRNA 体 外 酶 切:spCas9 1μL、10XspCas9 buffer 2μL、gRNA(体外转录)50ng、酶切的dsDNA 50ng、ddH2O up to 20μL,充分混合后,37℃反应30min,加入3μL DNA Loading Buffer 混合后65℃煮5min,跑2%琼脂糖凝胶电泳检测,对比与两个对照gRNA 估算活性。
1.4 pUbi-atU6-gRNA-XTH33 质粒的构建及鉴定
反应体系:XTH-G3-Sense 5μL、XTH-G3-Anti 5μL、ddH2O 15μL 混匀后,95℃3min,95℃到25℃缓慢冷却,并16℃5min。第一步 的 产 物1μL、Cas9/gRNAVector 1μL、Solution 1μL、Solution2 1μL、H2O 6μL 混匀后16℃反应2h。取第二步产物5~10μL 加入到刚解冻的50μLDH5a 感受态细胞中,混匀,冰浴30min 后,42℃热激90s,冰上静置2min,然后加入500μL 无抗LB,置于37℃恒温摇床中,170 转,复苏1h 后涂卡纳抗性的平板,培养过夜,挑取单菌落摇菌,并提取质粒,直接进行基因测序鉴定。鉴定正确的质粒与农杆菌GV3101 感受态一管,混合后冰浴10min。然后置于液氮中8min,随后37℃水浴5min。从水浴锅中取出后冰浴2~3min,加入800μl 无抗生素的YEB 液体培养基,28℃180rpm 摇床上培养4h 左右,取出500μl 菌液,均匀地涂布于加50mg/L Kan和50mg/L Rif 的YEB 琼脂培养基上,28℃培养出单菌落,并进行PCR 鉴定。
2 结果
2.1 体外gRNA 靶点活性检测
体外gRNA 靶点活性检测结果见图1,凝胶电泳结果显示G3 靶点活性明显大于标准品一的酶切效率,在50%以上,表明G3 靶点活性良好,可以使用。
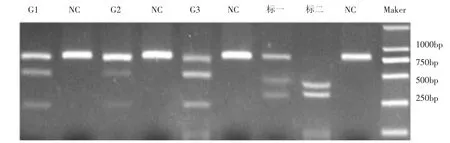
图1 体外gRNA 活性检测结果
2.2 pUbi-atU6-gRNA-XTH33 质粒构建结果
挑取2 个白色菌落摇菌,提质粒进行测序鉴定,测序比对结果见图2。结果显示靶点序列全部插入正确,XTH33 敲除质粒pUbi-atU6-gRNA-XTH33 构建成功。
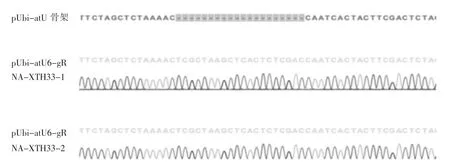
图2 质粒测序比对结果
2.3 pUbi-atU6-gRNA-XTH33 质粒转入农杆菌
质粒转入农杆菌感受态GV3101 后,28℃培养24h,出单菌落,挑取6 个单菌落,用引物XTH-G3-Sense:TTGGTCGAGAGTGAGCTTAGCGAGGG,pUbi-atU6-RP:GATGAAGTGGACGGAAG GAAGGAG 进行PCR 鉴定。结果显示1、2 号菌落批出430bp 的目的条带,鉴定正确。成功将pUbi-atU6-gRNA-XTH33 质粒转入农杆菌GV3101,鉴定结果见图3。
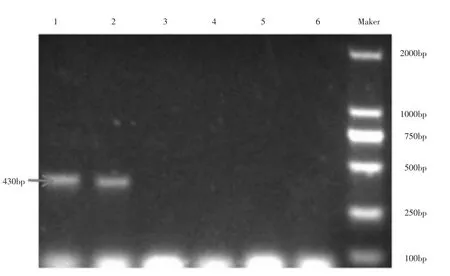
图3 农杆菌菌液PCR 鉴定结果
3 讨论
CRISPR 系统是由Cas 基因和成簇间隔的短回文重复序列组成。其原理是gRNA 序列可以靶向识别可以碱基互补的DNA 序列,并在Cas9 蛋白作用下特异性地切割其靶序列[2]。与传统的siRNA 和shRNA 基因沉默技术相比,CRISPR/Cas9 技术直接在基因水平进行操作,改变基因组成,使基因沉默效果更加彻底,而且CRISPR/Cas9 敲除质粒构建更为简单和高效,对于同物种不同基因,只需要设计不同基因的gRNA 序列,并插入载体骨架即可,但Cas9/gRNA 剪切活性与gRNA 靶点的识别序列有关,gRNA 的活性高低直接影响基因剪切效率。因此,灵活高效地选取高活性的gRNA,是进行敲除质粒构建并进一步实现基因编辑的关键。
CRISPR/Cas9 系统的载体构建中,为了方便高效的选取高活性gRNA 序列,提高基因敲除成功率,实验中首先设计3 条靶向拟南芥XTH33 基因的gRNA,通过体外检测手段检测3 条gRNA活性,选取活性最高的靶点XTH33-G3,靶点活性在50%以上,用活性最高的G3 靶点构建敲除质粒pUbi-atU6-gRNA-XTH33。通过体外gRNA 活性活性检测,可以初步判定gRNA 的效率,以便得到高效的敲除质粒,此方法省时省力,加快了实验进度,减少了试错机会,可以节省不必要的费用,并提高了基因敲除成功率。
本实验,成功进行了体外gRNA 活性检测,并构建XTH33 基因的CRISPR/Cas9 高效敲除质粒,为以后高效选取gRNA 提供了借鉴,并为下一步进行XTH33 基因功能鉴定奠定基础。
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
力学与实践(2022年3期)2022-07-02
现代食品(2022年6期)2022-04-19
复旦学报(医学版)(2021年4期)2021-08-05
江西农业学报(2021年4期)2021-04-20
农业工程学报(2020年20期)2020-12-25
矿产勘查(2020年11期)2020-12-25
航空发动机(2020年3期)2020-07-24
三农资讯半月报(2020年11期)2020-06-21
