
鞣花酸磷脂复合物的制备及其口服生物利用度研究
2021-09-27 09:37张留超刘勇华
中成药 2021年7期
张留超,刘勇华
(黄河科技学院,河南 郑州 450005)
文献[5⁃7] 报道,药物与磷脂形成磷脂复合物后其水溶性、脂溶性均可得到提高,有助于促进药物口服吸收;蒋新龙等[8]采用质子溶剂乙醇作为反应溶剂制备了鞣花酸磷脂复合物,但其复合率不足92%,可能与溶剂选择不当、工艺考察不精确有关。本实验以非质子溶剂四氢呋喃为反应溶剂[9]制备鞣花酸磷脂复合物,并考察其口服生物利用度,以期为相关制剂研究提供参考。
1 材料
Agilent 1200 型高效液相色谱仪(美国Agilent公司);SH23⁃1 型磁力搅拌器(上海江星仪器有限公司);AR1140 型电子天平[梅特勒⁃托利多仪器(上海)有限公司];DZF⁃6050 型真空干燥箱(上海欣齐科学仪器有限公司);XPert Powder 型X 射线衍射仪(英国马尔文公司);RE⁃3000 型旋转蒸发仪(上海亚荣生化仪器厂)。
鞣花酸原料药(纯度98.0%,批号190320,武汉荆楚陈氏医药化工有限公司);鞣花酸对照品(纯度99.6%,批号110726⁃201707,中国食品药品检定研究院);大豆卵磷脂(批号PC98T,磷脂酰胆碱质量分数>98%,上海辅必成医药科技有限公司)。清洁级SD 大鼠,体质量180~220 g,购自河南省实验动物中心,动物生产许可证号SCXK(豫)2016⁃0012。
2 方法与结果
2.1 鞣花酸磷脂复合物制备 取50 mg 原料药(45 ℃下真空干燥1 d)、处方量磷脂,置于三角瓶中,加入四氢呋喃制得混悬液,立即密封,置于一定温度水浴中,磁力搅拌适当时间后得澄清溶液,真空旋蒸除去有机溶剂,即得(性状为浅黄色半固体)。
2.2 鞣花酸含量测定
2.2.1 色谱条件 Waters Extend⁃C18色谱柱(150 mm×4.6 mm,5 μm);检测波长254 nm;流动相甲醇⁃水(含0.1%磷酸)(45 ∶55);体积流量1.0 mL/min;柱温35 ℃;进样量20 μL。
2.2.2 线性关系考察 精密称取鞣花酸对照品10.0 mg,溶于1.0 mL 四氢呋喃中,甲醇定容至10 mL,得1.0 mg/mL 母液,精密吸取1.0 mL 至100 mL量瓶中,流动相定容至刻度,得到10.0 μg/mL对照品溶液,“2.2.1”项下流动相稀释至10.0、5.0、2.5、0.5、0.1、0.05 μg/mL,在“2.2.1”项色谱条件下进样测定。以峰面积(Y)对鞣花酸质量浓度(X)进行回归,得方程为Y=14.314 2X+0.648 9(r=0.999 8),在0.05~10.0 μg/mL 范围内线性关系良好。
2.2.3 供试品溶液制备 取鞣花酸磷脂复合物约10 mg,置于100 mL 量瓶中,甲醇定容至刻度,取2.5 mL 至10 mL 量瓶中,“2.2.1”项下流动相定容至刻度,即得。
1.2 标本采集 抽取孕妇空腹外周静脉血5 ml装入真空普管中。待自凝后取血清3 000 r/min低温离心5 min,吸取上清液,置于-22°冰箱保存待同批测量。
2.2.4 方法学考察 取鞣花酸磷脂复合物适量,按“2.2.3”项下方法平行制备6 份供试品溶液,在“2.2.1”项色谱条件下进样测定,测得鞣花酸峰面积RSD 为1.20%,表明该方法重复性良好。取同一份供试品溶液,于0、2、4、8、12、24 h在“2.2.1”项色谱条件下进样测定,测得鞣花酸峰面积RSD 为0.58%,表明溶液在24 h 内稳定性良好。取“2.2.2”项下0.5、2.5、10.0 μg/mL对照品溶液,在“2.2.1”项色谱条件下进样测定6 次,测得鞣花酸峰面积RSD 分别为0.62%、0.43%、0.16%,表明仪器精密度良好。称取9 份鞣花酸磷脂复合物,加入“2.2.2”项下1.0 mg/mL对照品溶液1.25、2.50、3.75 mL,在“2.2.3”项色谱条件下进样测定,测得鞣花酸平均加样回收率分别为98.96%、100.68%、99.17%,RSD 分别为1.11%、0.84%、1.50%。
2.3 复合率测定 精密称取一定量鞣花酸(X0)制备磷脂复合物,加入10 mL 乙醚,振荡溶解后过0.22 μm 微孔滤膜,滤液减压旋蒸除去乙醚,收集固体至100 mL 量瓶中,甲醇定容至刻度,取2.5 mL至10 mL 量瓶中,“2.2.1”项下流动相定容,在“2.2.3”项色谱条件下进样测定参加复合的鞣花酸质量(X1),计算复合率,公式为复合率=(X1/X0)×100%。
2.4 Box⁃Behnken 响应面法 固定鞣花酸用量不变,课题组前期发现磷脂用量(A)、制备温度(B)、制备时间(C)是影响复合率的主要影响因素,三者水平见表1。再选择复合率(Y)作为评价指标,采用Box⁃Behnken 响应面法优化制备工艺,结果见表2。

表1 因素水平Tab.1 Factors and levels
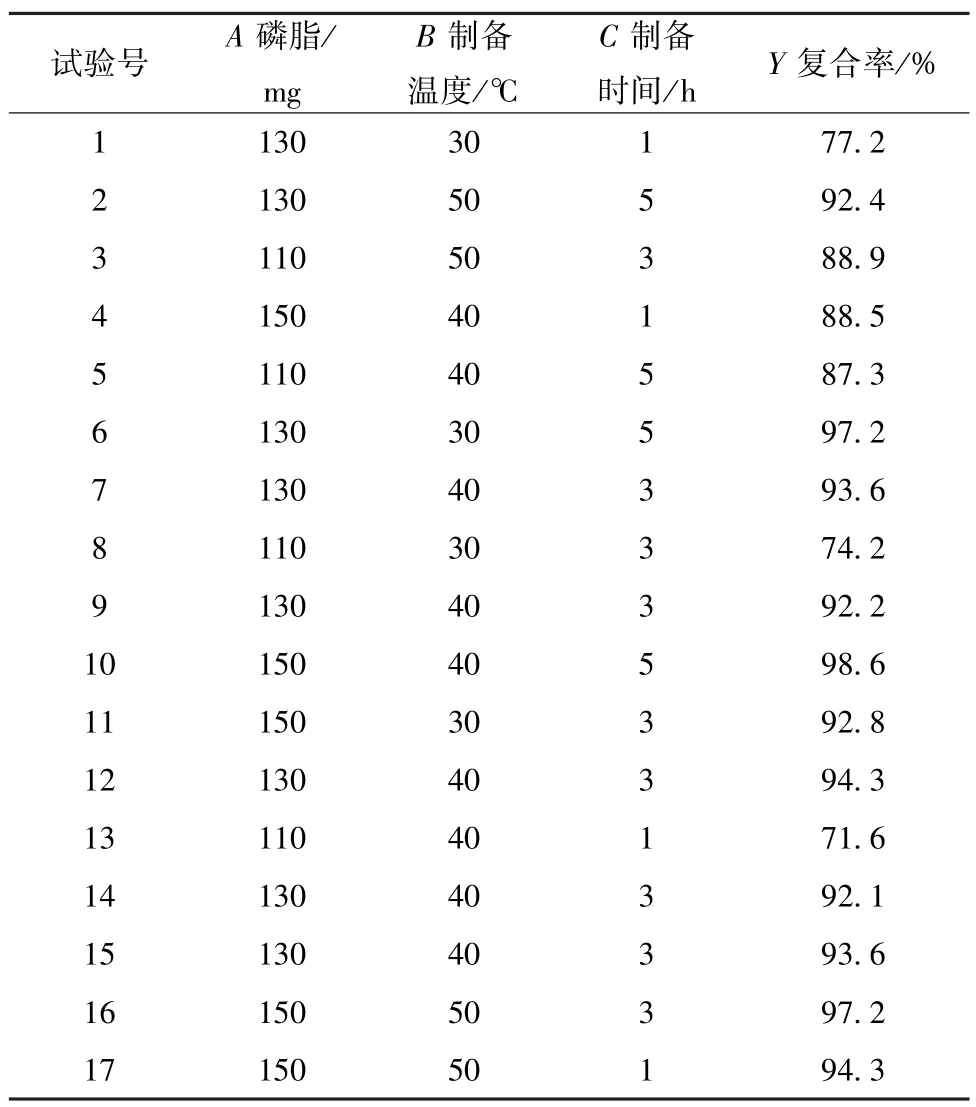
表2 试验设计与结果Tab.2 Design and results of tests
对表2 数据进行拟合,得到二次多元回归方程为Y=93.16 +6.89A+3.90B+5.51C- 2.58AB-1.40AC-5.47BC-4.33A2-0.55B2-2.33C2(R2=0.983 3,P<0.000 1,F=0.082 5,=0.961 9),R2较接近,表明拟合度良好;F>0.05,表明未知影响因素对实验结果干扰很小,模型可信度较高,方差分析见表3。由此可知,各因素对复合率均有显著影响(P<0.01),以A更明显,可能是由于磷脂用量不足会严重影响复合率所致,并且交互项AB、BC也有显著影响(P<0.05,P<0.01)。
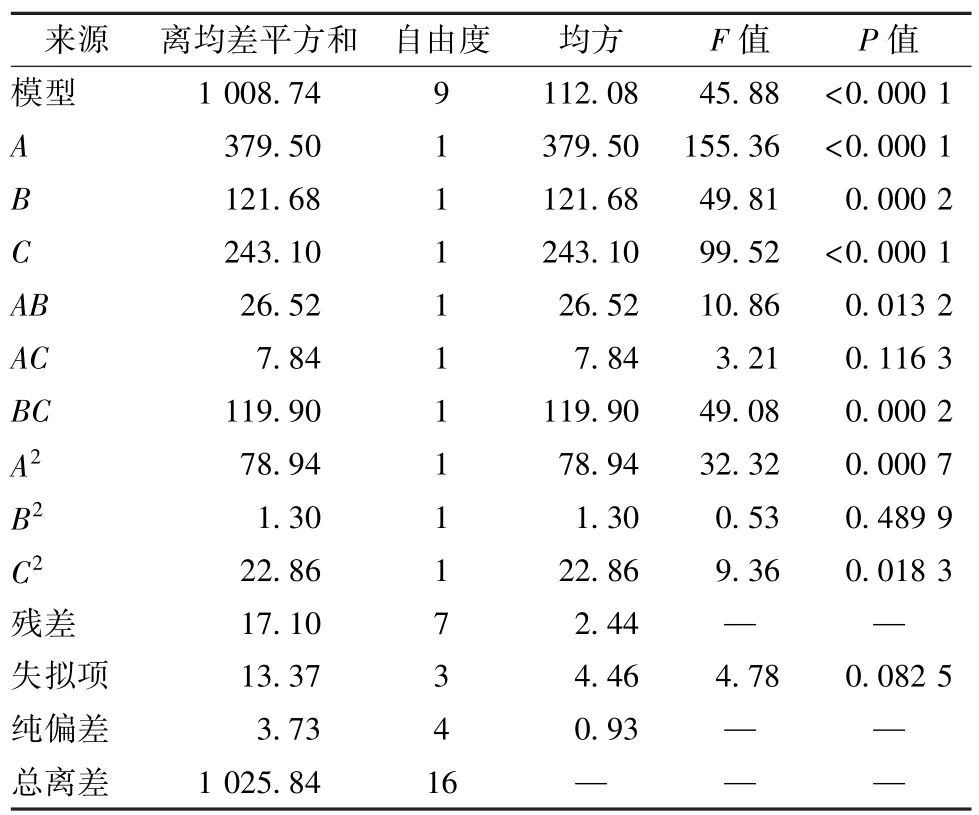
表3 方差分析Tab.3 Analysis of variance
通过Design Expert V8.0.6 软件进行分析,结果见图1。由图1a~1b 可知,固定某一因素时复合率随均随着另外2 个因素同时增加而增大,而后有所下降;由图1c 可知,虽然随着制备温度、制备时间增加复合率逐渐增大,但达到一定程度时反而明显下降,可能是过高的制备温度、过长的制备时间会对磷脂或其复合物稳定性产生一定影响,从而影响复合率。
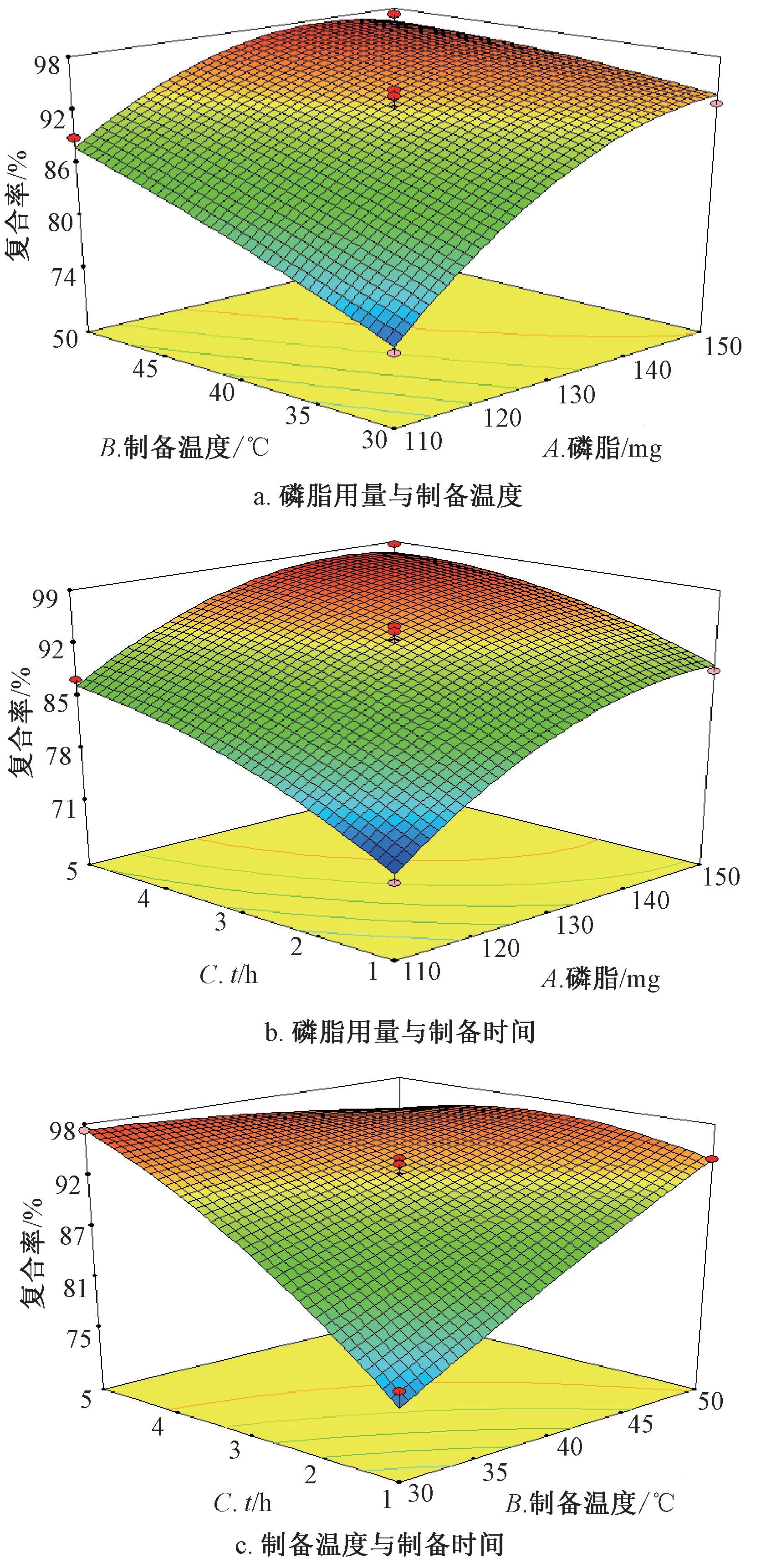
图1 各因素响应面图Fig.1 Response surface plots for various factors
设置复合率最大值为100%,最小值为0,得到最优制备工艺为磷脂用量141.6 mg(鞣花酸与磷脂比例约为1 ∶1.1),制备温度46.9 ℃,制备时间4.7 h,复合率为99.0%。按上述优化工艺制备3 份鞣花酸磷脂复合物,测得复合率分别为99.4%、99.7%、98.9%,平均99.3%,与预测值99.0%接近(相对误差为0.3%),表明模型预测性、重复性良好。
2.5 存在状态分析 XRPD 扫描条件为Cu⁃Kα 靶,扫描范围(2θ)4°~40°,电 流40 mA,速 度8°/min。取约10 mg 待测物,玻璃片压平后固定于X 射线粉末衍射仪上测定,结果见图2。由此可知,原料药在9.3°、10.1°、12.1°、28.4°等处出现特征晶型峰;在物理混合物(比例同磷脂复合物)中,磷脂、鞣花酸强度均发生较大幅度的下降,前者晶型衍射峰基本消失,但仍可发现后者在12.1°、28.4°处的特征晶型峰;磷脂复合物中鞣花酸所有晶型衍射峰均消失,表明它是不同于物理混合物的一种物质,鞣花酸在其中以无定型状态存在。
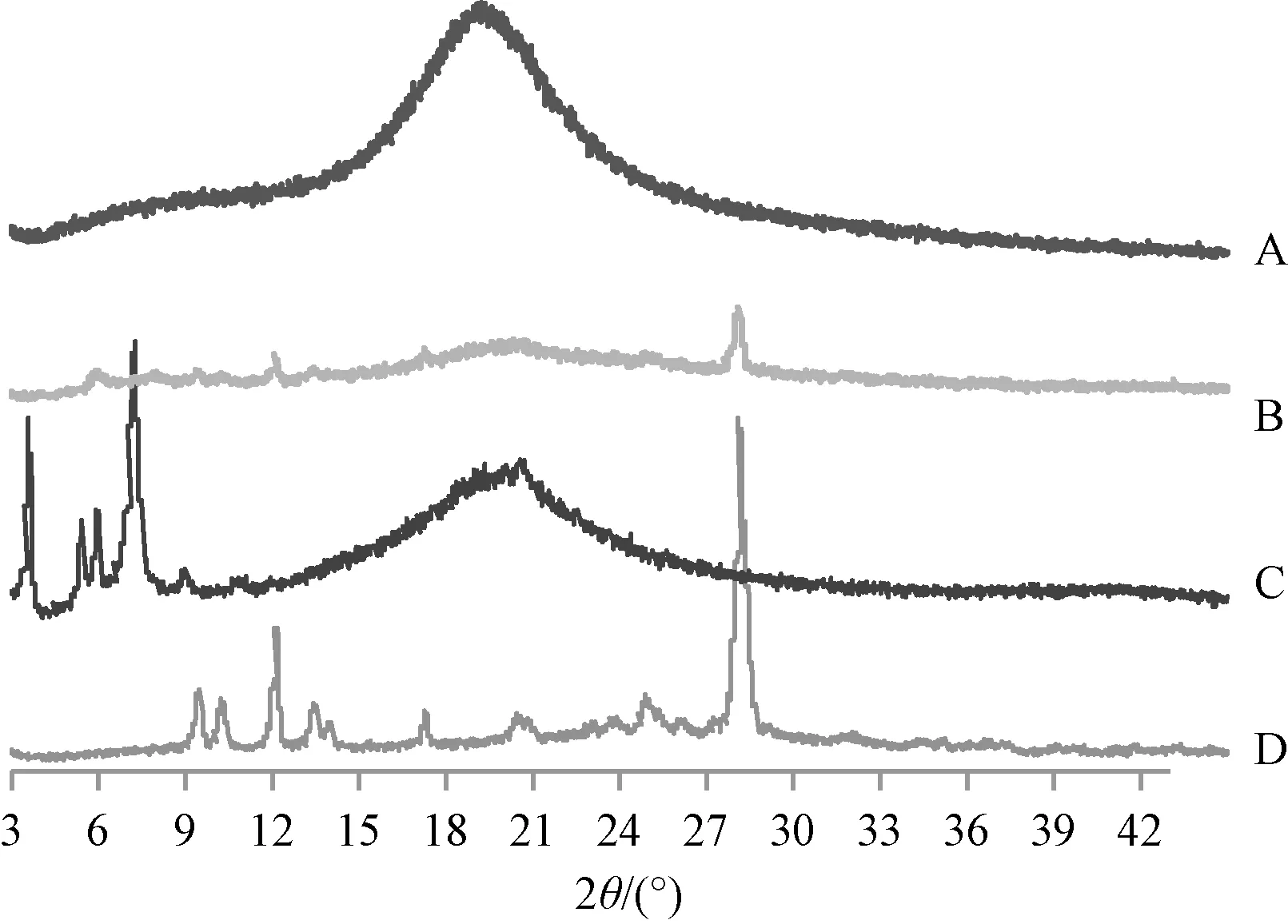
图2 样品XRPD 图Fig.2 XRPD patterns for samples
2.6 溶解度测定 取过量鞣花酸、物理混合物、磷脂复合物至三角瓶中,加入蒸馏水或正辛醇,25 ℃下磁力搅拌48 h,分别取3 mL 至离心管中,6 000 r/min 离心5 min,取上清液,测定表观溶解度,结果见表4,可知鞣花酸制成磷脂复合物后在水、正辛醇中的表观溶解度分别提高至3.22、7.18 倍。另外,该成分在物理混合物中的溶解度也有所提高,可能是由于磷脂增溶作用所致。
表4 样品表观溶解度测定结果(, n=6)Tab.4 Results of apparent solubility determination of samples(, n=6)

表4 样品表观溶解度测定结果(, n=6)Tab.4 Results of apparent solubility determination of samples(, n=6)
注:与鞣花酸比较,**P<0.01;与物理混合物比较,##P<0.01。
2.7 口服生物利用度研究
2.7.1 分组、给药与采血 取原料药、物理混合物、磷脂复合物适量,加入0.5% CMC⁃Na 溶液,超声30 s 后得混悬液(鞣花酸质量浓度均为15 mg/mL)。取过夜禁食的18 只大鼠,随机分为原料药组、物理混合物组、磷脂复合物组,每组6只,灌胃给药,剂量100 mg/kg,灌胃后立即计时,于0.15、0.25、0.75、1、1.5、2、3、4、6、8、12 h 将大鼠置于盛有乙醚的容器中麻醉10 s后,立即用浸润有肝素抗凝剂的玻璃毛细管眼眶采血各约0.2 mL,引流至涂有肝素抗凝的离心管中,3 000 r/min 离心2 min,血浆保存于-10 ℃冰箱中。
2.7.2 血浆样品处理 参考文献[3,10] 报道的方法,取100 μL 血浆样品,加入150 μL 1 mol/L KH2PO4、20 μL 50%磷酸,涡旋混合5 min,加入1 mL 乙腈涡旋混合5 min,7 500 r/min 离心5 min,转移上清液,再加入0.5 mL 乙腈洗涤,合并2 次上清液,N2缓慢吹干,于残渣中加入100 μL 甲醇,超声10 s,7 500 r/min 离心5 min。
2.7.3 线性关系考察 取鞣花酸对照品适量,甲醇制成1 000、500、250、100、50、25 ng/mL 对照品溶液,分别取100 μL 至离心管中,氮气缓慢吹干得残渣,加入100 μL 空白血浆,涡旋5 min,即得1 000、500、250、100、50、25 ng/mL 血浆对照品溶液,按“2.7.2”项下方法处理,在“2.2.1”项色谱条件下进样测定。以峰面积为纵坐标(Y),鞣花酸质量浓度为横坐标(X)进行回归,得方程Y=0.012 5X+0.055 3(r=0.993 8),在25~1 000 ng/mL 范围内线性关系良好。
2.7.4 方法学考察 取空白血浆、含药血浆(磷脂复合物给药12 h)、空白血浆+25 ng/mL 血浆对照品溶液,按“2.7.2”项下方 法处理,在“2.2.1”项色谱条件下进样测定,结果见图3,可知鞣花酸出峰时间为9.6 min,血浆内源性物质不干扰测定,专属性强。室温下于0、4、8、12、24、48 h 在“2.2.1”项色谱条件下进样测定,测得鞣花酸峰面积RSD 为6.39%,表明样品在48 h 内稳定性良好。取“2.7.3”项 下25、250、1 000 ng/mL血浆对照品溶液,在“2.2.1”项色谱条件下进样测定6 次,测得鞣花酸峰面积分别为3.14%、4.04%、1.68%,表明仪器精密度良好。取鞣花酸对照品适量,按“2.7.3”项下方法制备25、250、1 000 ng/mL 血浆对照品溶液,按“2.7.2”项下方法处理,在“2.2.1”项色谱条件下进样测定,测得平均加样回收率分别为90.43%、87.69%、92.05%,RSD 分别为4.07%、3.22%、2.48%。将“2.7.3”项下25 ng/mL 血浆对照品溶液逐步稀释,测得定量限(S/N=10)为5.0 ng/mL,检测限(S/N=3)为2.0 ng/mL。
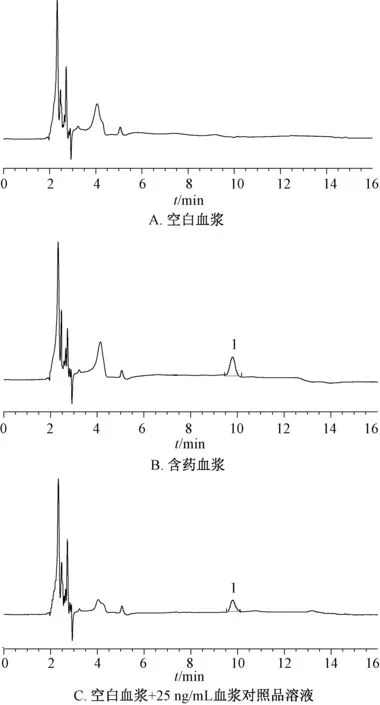
图3 鞣花酸HPLC 色谱图Fig.3 HPLC chromatograms of ellagic acid
2.7.5 药动学研究 图4、表5 显示,与原料药、物理混合物比较,磷脂复合物tmax缩短(P<0.05),Cmax、AUC0~t、AUC0~∞升高(P<0.01),相对生物利用度提高至2.46 倍。
表5 鞣花酸主要药动学参数(, n=6)Tab.5 Main pharmacokinetic parameters for ellagic acid(, n=6)

表5 鞣花酸主要药动学参数(, n=6)Tab.5 Main pharmacokinetic parameters for ellagic acid(, n=6)
注:与原料药比较,*P<0.05,**P<0.01;与物理混合物比较,#P<0.05,##P<0.01。
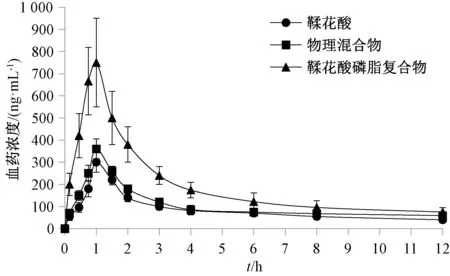
图4 鞣花酸血药浓度⁃时间曲线Fig.4 Plasma concentration⁃time curves for ellagic acid
3 讨论
本实验选择非质子溶剂(四氢呋喃)制备鞣花酸磷脂复合物,其复合率高于质子溶剂(如乙醇等)制备者,可能是质子溶剂会对药物和磷脂形成复合物时产生一定干扰作用,从而影响了复合率。所得最优制备工艺的复合率接近100%,而且鞣花酸在水、正辛醇中的溶解度分别提高至3.22、7.18 倍,远高于乙醇制备者(1.4、2.1 倍)[8],具体原因有待进一步研究。
在血浆样品处理过程中,药物损失会影响含量测定的准确性,一般采用内标法。但对于鞣花酸血浆样品而言,在酸性条件下采用外标法[3,10⁃12]处理时,血浆内源性物质不干扰测定,回收率均大于87.69%,而且操作简单,故本实验也选择该方法,与Mady 等[3]报道基本一致,可能是由于在酸性环境中更有利于鞣花酸进入乙腈而被有效提取出。
药动学结果显示,磷脂复合物可将鞣花酸口服吸收生物利用度提高至2.50 倍,可能与该剂型改变了后者存在状态、提高了其水溶性和脂溶性有关,但该成分易被胃肠道酶解[11]。文献[13] 报道,磷脂复合物提高药物生物利用度还与灌胃液配制方式有关,并且其程度也可能存在差异。本实验所制备的鞣花酸磷脂复合物脂溶性得到极大改善,可为后续相关纳米制剂的开发奠定基础[14⁃17]。
猜你喜欢
纺织标准与质量(2022年3期)2022-08-10
承德医学院学报(2022年2期)2022-05-23
中成药(2019年12期)2020-01-04
中成药(2018年7期)2018-08-04
中成药(2018年6期)2018-07-11
中成药(2017年12期)2018-01-19
中成药(2017年5期)2017-06-13
中成药(2016年8期)2016-05-17
中国民族医药杂志(2016年5期)2016-05-09
中国民族医药杂志(2016年4期)2016-05-09
