
二茂铁基-双酮锌配合物的合成、电化学活性及多光子吸收
2021-09-22 02:12:30解清园李丹丹李胜利吴杰颖田玉鹏
无机化学学报 2021年9期
解清园 李 丹 李丹丹 李胜利 吴杰颖 张 琼*,,2 田玉鹏*,,2
(1安徽大学化学系,功能无机材料化学安徽省重点实验室,合肥 230601)
(2南京大学配位化学国家重点实验室,南京 210023)
二茂铁(ferrocene,Fc)的发现被认为是现代有机金属化学的起点[1‑2],Fc及其衍生物在催化[3‑4]、医药[5‑7]以及光、电、磁功能材料[8‑10]等许多领域都有极广泛的应用[11]。Fc的2个环戊二烯基负离子都具有芳香性,容易发生亲电取代反应、锂化反应以及还原反应,可以通过与多种功能性基团结合,形成结构类型多样和具有独特电化学及光学特性的Fc衍生物[12‑15]。此外,给体-受体型Fc衍生物易于发生分子内电荷转移,能有效地诱导体系的非对称极化,表现出优秀的非线性光学性质[16]。β‑双酮是一种重要的功能性吸电子基团,其在失去质子后作为双齿配体与金属离子配位形成共轭单元[17]。当β‑双酮衍生物失去一个质子作为配体与Zn2+进行配位时,能够得到具有良好的化学稳定性、电子离域程度大的非线性光学效应配合物[18]。根据近期文献调研,具有三光子吸收效应的二茂铁β‑双酮锌配合物尚未见报道。
我们经两步反应合成了具有D‑A构型的功能性配体β‑双酮二茂铁(L)及其锌配合物(NHEt3)[Zn(L)3],通过红外光谱、质谱、氢谱、元素分析和单晶解析等手段对其进行了表征。单晶解析发现锌配合物(NHEt3)[Zn(L)3]中的L以-1价的烯醇式结构与Zn2+配位,呈负电性,分子外围存在1个质子化的三乙胺分子,带1个正电荷,以中和配合物的负电性,使得整个分子呈电中性。电化学等性质研究表明,配体和配合物都具有较好的氧化还原可逆性。Fc、β‑双酮和中心金属离子之间显示出良好的电子离域作用,形成大的共轭体系,显示出明显的三光子吸收效应。
1 实验部分
1.1 试剂与仪器
所用试剂有Fc、乙酸酐、磷酸、氢氧化钾、三氟乙酸乙酯、二水合乙酸锌、三乙胺、乙醇、金属钠、石油醚、乙酸乙酯、甲醇、三乙胺、浓盐酸,均为分析纯,使用前按标准方法进行纯化。
1H NMR在Bruker Avance 400 MHz光谱仪上记录。红外光谱使用Nicolet FT‑IR 870 SX红外光谱仪(KBr压片)记录。紫外可见吸收光谱使用日立U‑3900紫外可见分光光度计测定。质谱数据使用Micro‑mass GCT‑MS 记录(ESI源)。在Bruker APEX‑Ⅱ CCD X 射线衍射仪(λ=0.071 069 nm,296 K)和Stoe Stadivari衍射仪(λ=0.154 186 nm,296 K)上对样品进行单晶X射线衍射数据采集。用飞秒激光脉冲钛宝石系统(680~1 080 nm,80 MHz,140 fs)测定双光子吸收性质。使用飞秒激光脉冲钛宝石光学参量振荡器系统(Chameleon Compact OPO,1 100~1 600 nm,140 fs)测定三光子吸收性质。电导率测试仪器为DDS‑307A电导仪。热重测试仪器为TGA5500R热重分析仪。电化学测试仪器为电化学分析仪CHI630(Shanghai Chenhua Instrument),实验是在三电极体系中进行的,以铂盘电极作为工作电极,铂丝电极作为对电极,Ag/AgCl电极作为参比电极,高氯酸四丁基铵(TBAP)作为支持电解质,浓度为0.1 mol·L-1,扫描电压范围:0~1.2 V,测试用溶剂为乙腈,测试溶液浓度为1.0 mmol·L-1。原位红外电化学实验采用自制薄层池作为红外光谱电化学池,使用的红外光谱仪为Nicolet IS50(Thermo Nicolet Corpra‑tion),检测器为 HgCdTe/A(MCT/A),使 用 Grans/3D software对数据进行处理。
1.2 实验过程
化合物合成路线如图1所示。
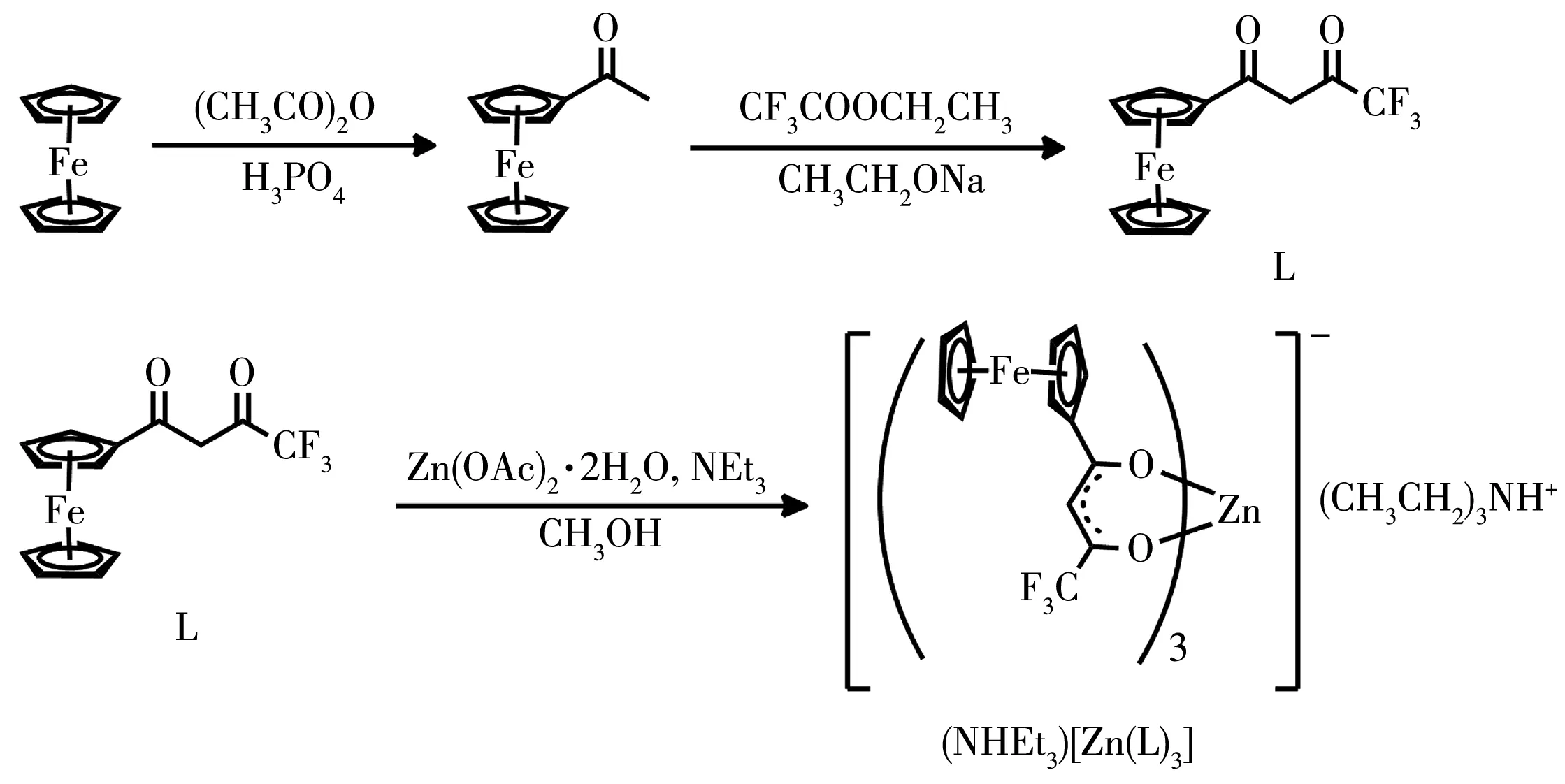
图1 配合物(NHEt3)[Zn(L)3]的合成路线Fig.1 Synthesis route of complex(NHEt3)[Zn(L)3]
1.2.1 配体L的合成
合成步骤参照文献[19],得紫红色产物8.3 g,产率为65%。m.p.363~365 K。MALDI‑TOF MS:m/z,计算值:324.07,实测值:323.06[M-H]-。IR(KBr,cm-1):3 415(s),3 093(s),1 654(s),1 597(s),1 478(m),1 302(s),1 192(s),1 147(s),1 001(s),823(m),678(m),510(m)。1H NMR(400 MHz,CD3COCD3):δ 6.21(s,1H),4.96(s,2H),4.66(s,2H),4.24(s,5H)。 元 素 分 析 按C14H11F3FeO2的计算值(%):C 51.89,H 3.42;实测值(%):C 51.78,H 3.39。
1.2.2 配合物(NHEt3)[Zn(L)3]的合成
称取1.037 0 g(3.2 mmol)二茂铁双酮配体L和0.496 4 g(4.90 mmol)三乙胺在20 mL甲醇溶液中常温搅拌10 min。称取0.219 4 g(1.0 mmol)Zn(OAc)2·2H2O溶于30 mL甲醇中,逐滴加入到上述三乙胺溶液中,回流2 h。冷却至室温,抽滤,得到黑红色片状微晶0.35 g(0.34 mmol),产率68%。m.p.476~480 K。IR(KBr,cm-1):3 423(w),2 930(w),1 609(s),1 529(s),1 438(s),1 378(s),1 294(s),1 132(s),1 092(m),1 029(m),941(m),826(m),790(m),674(m),500(s)。1H NMR(400 MHz,CDCl3):δ 5.93(s,3H),4.79(t,J=2.0 Hz,6H),4.44(t,J=2.0 Hz,6H),4.20(s,15H),3.06(d,J=8.6 Hz,6H),1.19(t,J=7.2 Hz,9H)。元素分析按 C48H46F9Fe3NO6Zn的计算值(%):C 50.71,H 4.08,N 1.23;实测值(%):C 51.01,H 4.083,N 0.991。
1.2.3 化合物的单晶X射线衍射分析
将适量配体L溶解于精制二氯甲烷中,过滤至25 mL的锥形瓶中,上层覆盖适量的乙醇或正己烷,缓慢挥发数天,得到可用于测试的黑色块状晶体。挑选配体L(0.300 mm×0.300 mm×0.300 mm)进行单晶衍射分析。衍射数据在Bruker APEX‑ⅡCCD X射线衍射仪上收集,用Mo靶(Kα射线,λ=0.071 069 nm)收集衍射数据,吸收校正采用多扫描方法。分子结构中的非氢原子采用直接法由Olex 2[20]和ShelXT[21]程序解出,使用ShelXL[22]对结构精修。氢原子为理论加氢,即氢原子的位置由几何构型计算得到。
将适量配合物(NHEt3)[Zn(L)3]溶解于精制甲醇中,过滤至25 mL的锥形瓶中,缓慢挥发数天,得到可用于测试的黑色块状单晶。挑选配合物(NHEt3)[Zn(L)3](0.210 mm×0.200 mm×0.200 mm)进行单晶衍射分析。衍射数据在Stoe Stadivari X射线衍射仪上收集,在296 K温度下,用Cu靶(Kα射线,λ=0.154 186 nm)收集衍射数据,采用ω‑2θ扫描方式收集衍射数据。使用Olex 2和ShelXT程序对结构进行解析,使用ShelXL对结构精修。氢原子为理论加氢,即氢原子的位置由几何构型计算得到。
CCDC:2070926,L;2070927,(NHEt3)[Zn(L)3]。
2 结果与讨论
2.1 晶体结构解析
由单晶X射线衍射数据得到的晶体结构图如图2和图3所示,相关晶体参数见表S1(Supporting in‑formation)。

图2 (a)L的30%椭球概率晶体结构图(省略H原子);(b)L的包含分子间氢键(用虚线表示)的三维结构Fig.2 (a)Molecular structure of L showing 30%probability displacement ellipsoids(all hydrogen atoms are omitted for clarity);(b)Three‑dimensional structure of L containing intermolecular hydrogen bonds(indicated by dotted lines)

图3 (NHEt3)[Zn(L)3]的30%椭球概率晶体结构图Fig.3 Molecular structure of(NHEt3)[Zn(L)3]showing 30%probability displacement ellipsoids
配体L的晶体结构如图2a所示,它属于单斜晶系,P1空间群。二茂铁基茂环和未取代茂环平面的距离分别为0.164 7和0.165 2 nm,含取代基茂环和未取代茂环的平面性很好,两平面的夹角为1.194(5)°,几乎平行。双酮的2个碳氧键O1—C11和O2—C13键长分别为0.127 7和0.128 9 nm,均介于正常的C—O单键(0.142 nm)和C=O双键(0.120 nm)之间,且更接近双键。双酮之间的桥键C11—C12和C12—C13键长分别为0.141 5和0.136 6 nm,双酮与茂环之间的桥键C10—C11键长为0.145 3 nm,均介于正常的C—C单键(0.153 nm)和C=C双键(0.132 nm)之间。分析可知,双酮的2个碳氧键O1—C11和O2—C13和双酮之间的桥键C11—C12和C12—C13存在大π键,以烯醇共振结构存在,且碳氧键O2—C13的键长大于O1—C11的键长,靠近三氟甲基吸电子基团的O2—C13键更易以烯醇式结构存在;茂环与双酮平面的平面性较好,且键长平均化,说明分子共轭程度高,电子离域性好,有利于双酮与Fc之间的电子交流。
L的三维结构如图2b所示。可以看到,L上的3个氟原子与同分子上的双酮间的氢原子形成氢键,与邻近的分子未取代茂环上的氢也形成氢键,并通过氢键作用形成三维结构。氢键的形成增大了其熔点,增强了热稳定性。此外,化合物L与极性溶剂分子也可能形成氢键,不仅能增大其在极性溶剂中的溶解度,也可能产生溶剂化效应。
配合物(NHEt3)[Zn(L)3]的晶体结构如图3所示,它属于单斜晶系,P21/n空间群。从图中可以看出,该配合物为单核结构,中心金属Zn2+与3个配体中的6个氧原子配位,形成八面体构型。在配合物中3个配体与中心Zn2+形成3个稳定的六元环(P1环:Zn1、O1、C11、C12、C13、O2;P2 环:Zn1、O3、C25、C26、C27、O4;P3 环:Zn1、O5、C39、C40、C41、O6)。其中,中心原子的配位键Zn—O键长在0.205 0~0.214 4 nm范围,平均键长为0.209 7 nm。配合物中6个碳氧键O1—C11、O2—C13、O3—C25、O4—C27、O5—39、O6—C41键长分别为 0.126 3、0.124 5、0.126 7、0.126 3、0.125 0、0.128 3 nm,均介于正常的C—O单键(0.142 nm)和C=O双键(0.120 nm)之间,平均键长为0.126 2 nm,与自由配体中的碳氧键的平均键长(0.128 3 nm)很接近,可以认为是双酮烯醇式的键长[23],表明3个配体的羟基上的质子离去,以-1价的烯醇式结构与Zn2+配位,整个分子呈负电性;并且配合物的碳氧键的平均键长小于自由配体的碳氧键的平均键长,说明配体和金属离子之间有较强的电子交流,并且相较于配体,在配体与锌配位后形成的配合物具有更好的电子离域性。
2.2 配合物的电导率
应用电导率仪在298 K温度下测定(NHEt3)[Zn(L)3]在乙腈溶剂中的电导率,根据Λm=(κ液-κ剂)/c公式求得(NHEt3)[Zn(L)3]的摩尔电导率为96.0 S·m2·mol-1,根据文献报道[24],可以确定该配合物电荷比为1∶1。
2.3 配合物的热稳定性
用热重(TG)法和微商热重(DTG)法研究化合物L和(NHEt3)[Zn(L)3]的热分解行为,测试条件:测试温度为15~800℃,N2氛围,升温速率10℃·min-1。如图4所示,TG曲线上10%的重量损失的分解温度分别为 155.8℃(L)和 178.2℃((NHEt3)[Zn(L)3])。在500℃时,配体及其配合物分别残留0%(L)和40%((NHEt3)[Zn(L)3]),并且配合物在211.8℃时,已经完全分解。TG和DTG曲线表明,配合物的热稳定性都远远超过配体L,这是因为配体L和锌配位后增强了稳定性。

图4 L和(NHEt3)[Zn(L)3]的TG(a)和DTG(b)曲线Fig.4 TG(a)and DTG curves(b)of L and(NHEt3)[Zn(L)3]
从TG曲线上进一步分析(NHEt3)[Zn(L)3]的热分解过程,可认为其分为2个阶段:第一阶段,温度范围为138.5~227.7℃,伴随有0%~32.6%的质量损失,可以认为是2个二茂铁基团的热分解;第二阶段,温度范围为245.4~398.9℃,伴随有34.3%~57.8%的质量损失,可以认为是第3个二茂铁基团和三乙胺的热分解。
2.4 配体与配合物的紫外可见吸收光谱性质
紫外可见吸收光谱与电子跃迁有关,分子内相应的电子吸收对应能量的光子将会由基态跃迁至激发态。通过吸收光谱可以了解到相应的分子结构和分子轨道的变化。L与(NHEt3)[Zn(L)3]分别在二氯甲烷中测定的紫外可见吸收光谱如图5所示。另外采用含时密度泛函理论(TD‑DFT)方法,在B3lYP/3‑21G水平上优化了配体和配合物的分子结构,计算得到配合物的分子轨道能级图(图6),理论计算的计算数据列在表1中。

图5 L和(NHEt3)[Zn(L)3]在二氯甲烷中的紫外可见吸收光谱Fig.5 UV‑Vis absorption spectra of L and(NHEt3)[Zn(L)3]in dichloromethane
从图5、图6和表1可以看出:配体L与配合物(NHEt3)[Zn(L)3]在350 nm附近均出现一个很强的吸收峰,可归属为配体与配合物上的分子内电荷迁移(ICT)过程,电子云从Fc的茂环转移到双酮基团,主要来自HOMO到LUMO+1的跃迁。
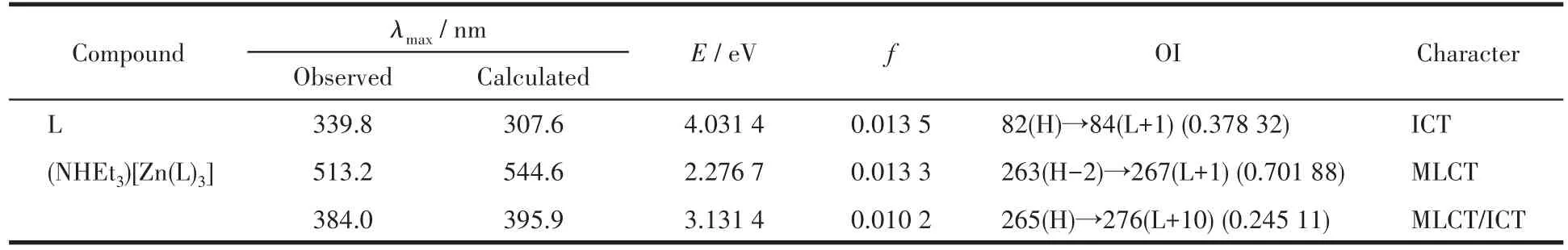
表1 L和(NHEt3)[Zn(L)3]的理论计算的吸收光谱数据*Table 1 Calculated absorption spectral properties of L and(NHEt3)[Zn(L)3]*
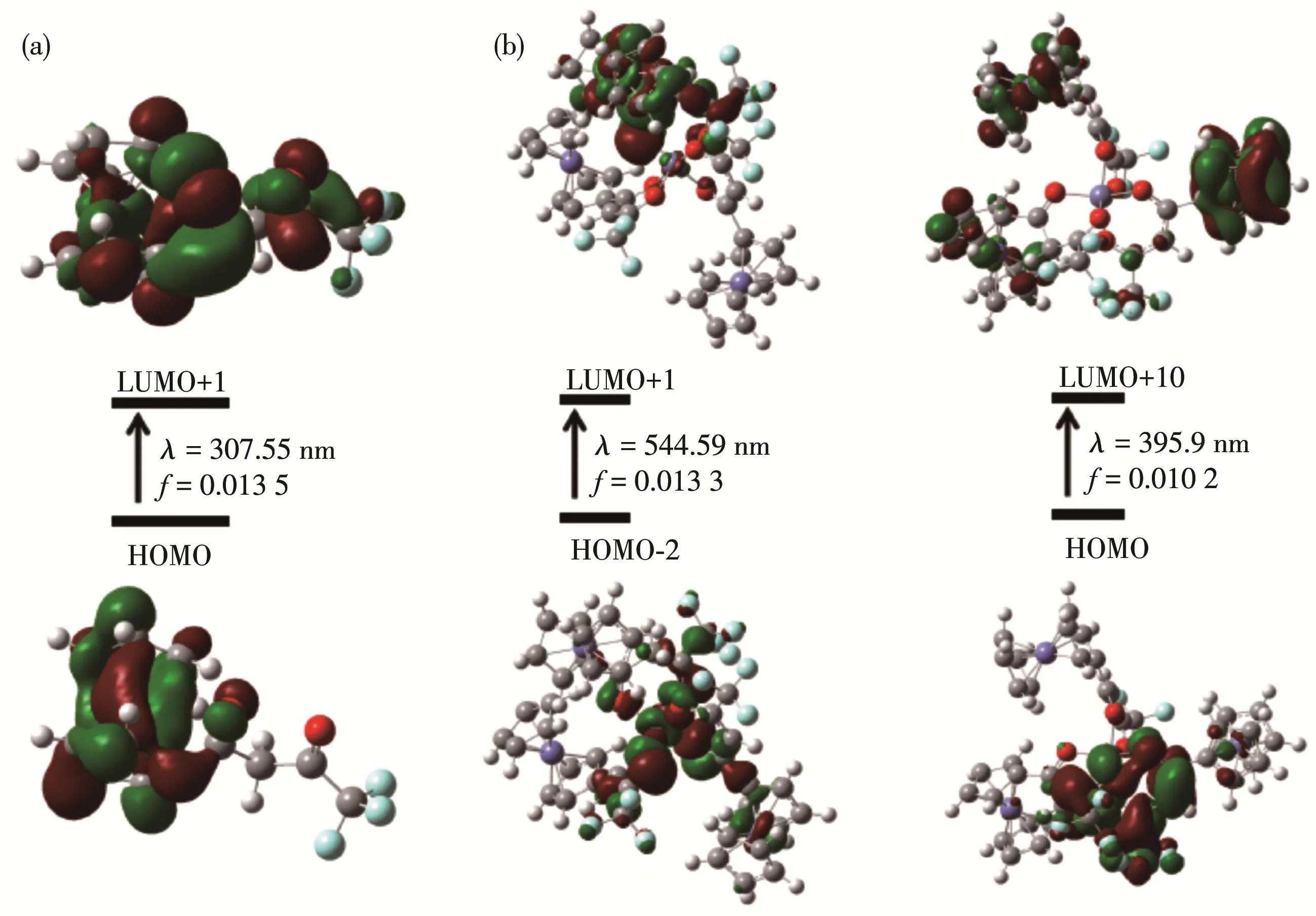
图6 化合物的能级图:(a)L、(b)(NHEt3)[Zn(L)3]Fig.6 Energy level diagram of the compounds:(a)L,(b)(NHEt3)[Zn(L)3]
配合物(NHEt3)[Zn(L)3]在513 nm处出现新峰,可以归属为金属锌中心与双酮配体间的电荷转移,结合理论计算可以确定锌配合物在荷移谱带的吸收峰为金属-配体电荷迁移(MLCT),电子云从锌中心转移到双酮配体,该带主要来自HOMO-2到LUMO+1的跃迁。
此外,(NHEt3)[Zn(L)3]位于383 nm处的吸收峰主要来自HOMO到LUMO+10的跃迁,结合理论计算可以确定在该处的吸收峰为MLCT和ICT,电子云从锌中心转移到Fc的茂环,伴有双酮基团到茂环的电荷转移。
2.5 配体与配合物的电化学性质
为了检测L及其锌配合物的电化学性质,以CH3CN为溶剂利用循环伏安(CV)法测试了Fc、L和(NHEt3)[Zn(L)3]的氧化还原特性,结果如图7和表2所示。
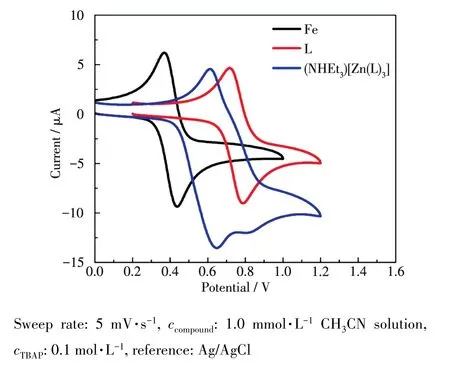
图7 Fc、L和(NHEt3)[Zn(L)3]的CV曲线Fig.7 CV curves of Fc,L and(NHEt3)[Zn(L)3]
从图7和表2可见,Fc、L以及(NHEt3)[Zn(L)3]均在0~1.2 V之间有氧化还原峰,可归结为Fc单元的氧化还原峰。通过对比发现,L的氧化还原峰同Fc相比发生了正移,氧化峰正移了0.325 V,还原峰正移了0.324 V。(NHEt3)[Zn(L)3]同Fc相比,其氧化还原峰位也发生明显正移;(NHEt3)[Zn(L)3]同L相比,氧化还原峰位发生了负移。这表明L和(NHEt3)[Zn(L)3]要比Fc更难被氧化,这可能是由于L和(NHEt3)[Zn(L)3]相较于Fc,共轭体系增大,电子离域性增强,二茂铁基团的电子云密度降低,被氧化能力降低。但(NHEt3)[Zn(L)3]相比于L更容易被氧化,并且L具有更好的氧化还原可逆性。这可能是由于锌离子金属中心具有d10电子结构,金属到配体的β‑双酮基团间的d‑p反馈键使得配合物的二茂铁基团上的电子云密度增强,较易发生氧化和还原。

表2 Fc、L和(NHEt3)[Zn(L)3]的CV数据*Table 2 CV data of Fc,L and(NHEt3)[Zn(L)3]*
从(NHEt3)[Zn(L)3]的循环伏安曲线不难看出,(NHEt3)[Zn(L)3]具有2个氧化峰和1个还原峰,结合晶体结构分析结果,推测该分子的3个Fc单元发生了分步氧化。(NHEt3)[Zn(L)3]的电子转移机理还可以通过快速扫描时间分辨FT‑IR光谱电化学(RS‑TRS FT‑IR)的3D光谱得到进一步验证。如图8a所示,可以观察到2个主要红外特征吸收峰(3 103、2 973 cm-1),这2个特征吸收峰在Fc及L的原位红外光谱中同样出现,它们可归因于化合物中二茂铁基的C—H伸缩振动,可以用来指认为化合物的氧化和还原的产物。如图8b所示,2 973 cm-1处的红外吸收峰在氧化过程中逐渐降低并且消失,在还原过程中,开始出现并达到最大值。3 103 cm-1处的吸收峰在氧化过程中开始增大,然后在还原过程中逐渐消失,该吸收峰可用来追踪Fc氧化产物(Fc+)[25]。
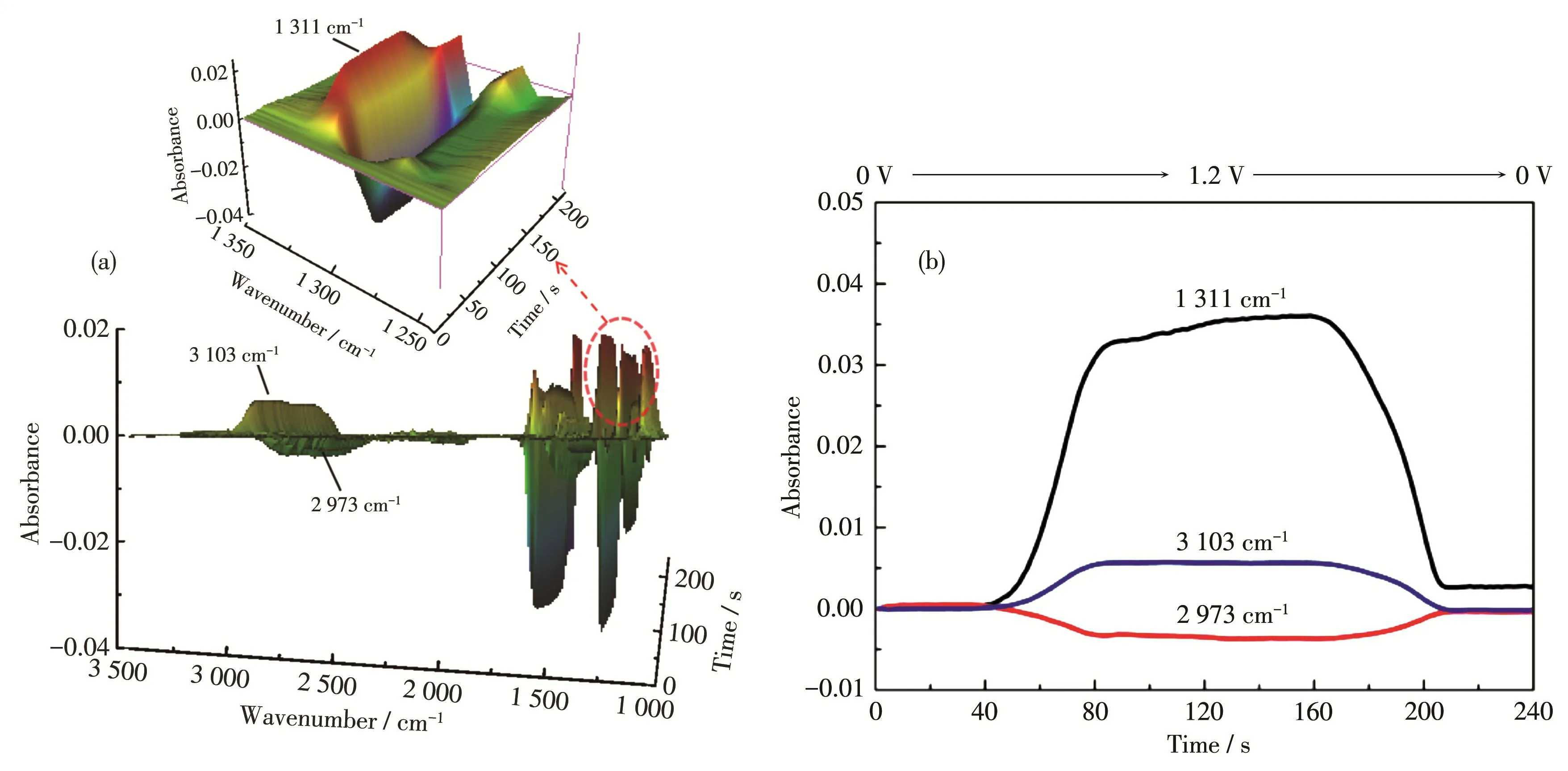
图8 (a)(NHEt3)[Zn(L)3]在电位扫描速率5 mV·s-1时的3D光谱(插图:虚线部分的放大图);(b)(NHEt3)[Zn(L)3]在1 311 cm-1(黑线)、2 973 cm-1(红线)和3 103 cm-1(蓝线)处电化学反应的循环伏安-吸收图(用快速傅里叶变换平滑算法平滑)Fig.8 (a)3D spectra of(NHEt3)[Zn(L)3]with potential scanning rate of 5 mV·s-1(Inset:enlarged view of the dotted line);(b)Cyclic voltammetry‑absorption diagram for electrochemical reaction of(NHEt3)[Zn(L)3]at 1 311 cm-1(black line),2 973 cm-1(red line)and 3 103 cm-1(blue line)(smoothed by fast Fourier transform smoothing algorithm)
可以认为(NHEt3)[Zn(L)3]在氧化之前由于3个二茂铁基团和锌中心都具有给电子能力,电子分布均匀,1 311 cm-1处的谱带不可见。但是,随着氧化过程中电位的增加,1个Fc单元(作为供体,D)被氧化成Fc+(作为受体,A)。Fc+单元具有吸电子能力,形成D‑A不对称结构模式,出现了1 311 cm-1吸收峰。随着氧化的继续,另外2个Fc也被氧化,3个Fc+与锌中心仍成D‑A结构模式。随着电势变小,Fc+还原成Fc,构型从D‑A变为D‑D,所以1 311 cm-1处吸收峰消失。这一电子传递机理如图9所示。
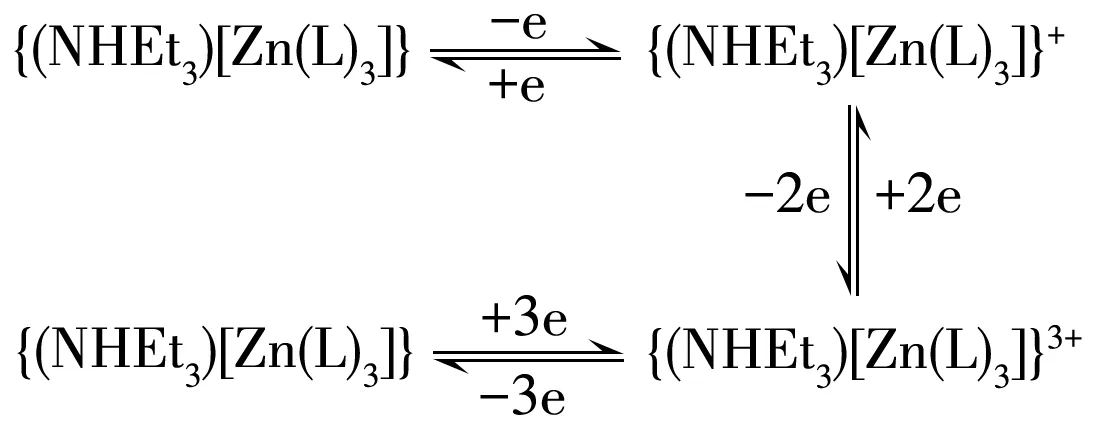
图9 (NHEt3)[Zn(L)3]的电子传递机理Fig.9 Electron transport mechanism of(NHEt3)[Zn(L)3]
2.6 配体与配合物的非线性光学性质
Fc衍生物通常具有较好的非线性光学性质,基于L和(NHEt3)[Zn(L)3]分子内有较强的电子流动,我们采用开孔Z‑scan方法测试了配体和配合物在近红外区(~850 nm和~1 300 nm)的非线性光学性质。图10是配体L和配合物(NHEt3)[Zn(L)3]的开孔/闭孔Z‑scan图,表3列出了L和(NHEt3)[Zn(L)3]的主要非线性光学数据。左景林等[26]通过Z‑scan方法探索了[Me4N][Au(dmit)2]和[nBu4N][Au(dmit)2]的三阶非线性光学性质,2个dmit配体的Au配合物χ(3)值分别为6.8×10-13和8.06×10-13esu。Saouma等[27]通过三次谐波产生(THG),探索了二维R‑P钙钛矿(CH3(CH2)3NH3)2(CH3NH3)n-1PbnI3n+1(n=1~4)的三阶非线性光学性质,χ(3)范围为(2.6±0.5)×10-11~(5.6±1.0)×10-11esu。我们制备的配体L和配合物(NHEt3)[Zn(L)3]的三阶非线性极化率远大于文献报道值。
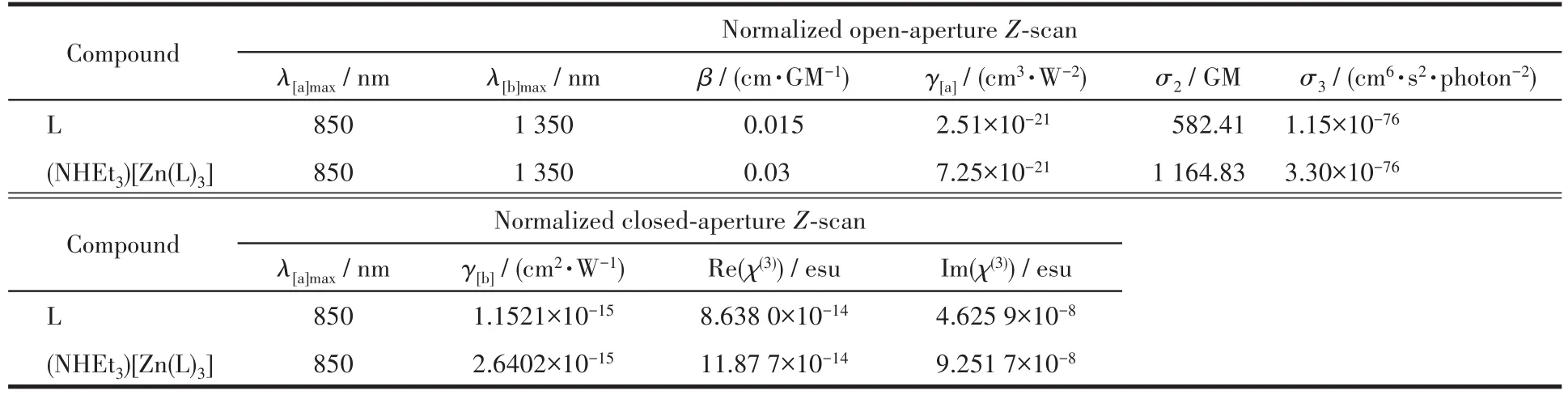
表3 L和(NHEt3)[Zn(L)3]的多光子吸收数据*Table 3 Multi⁃photon absorption data of L and(NHEt3)[Zn(L)3]*
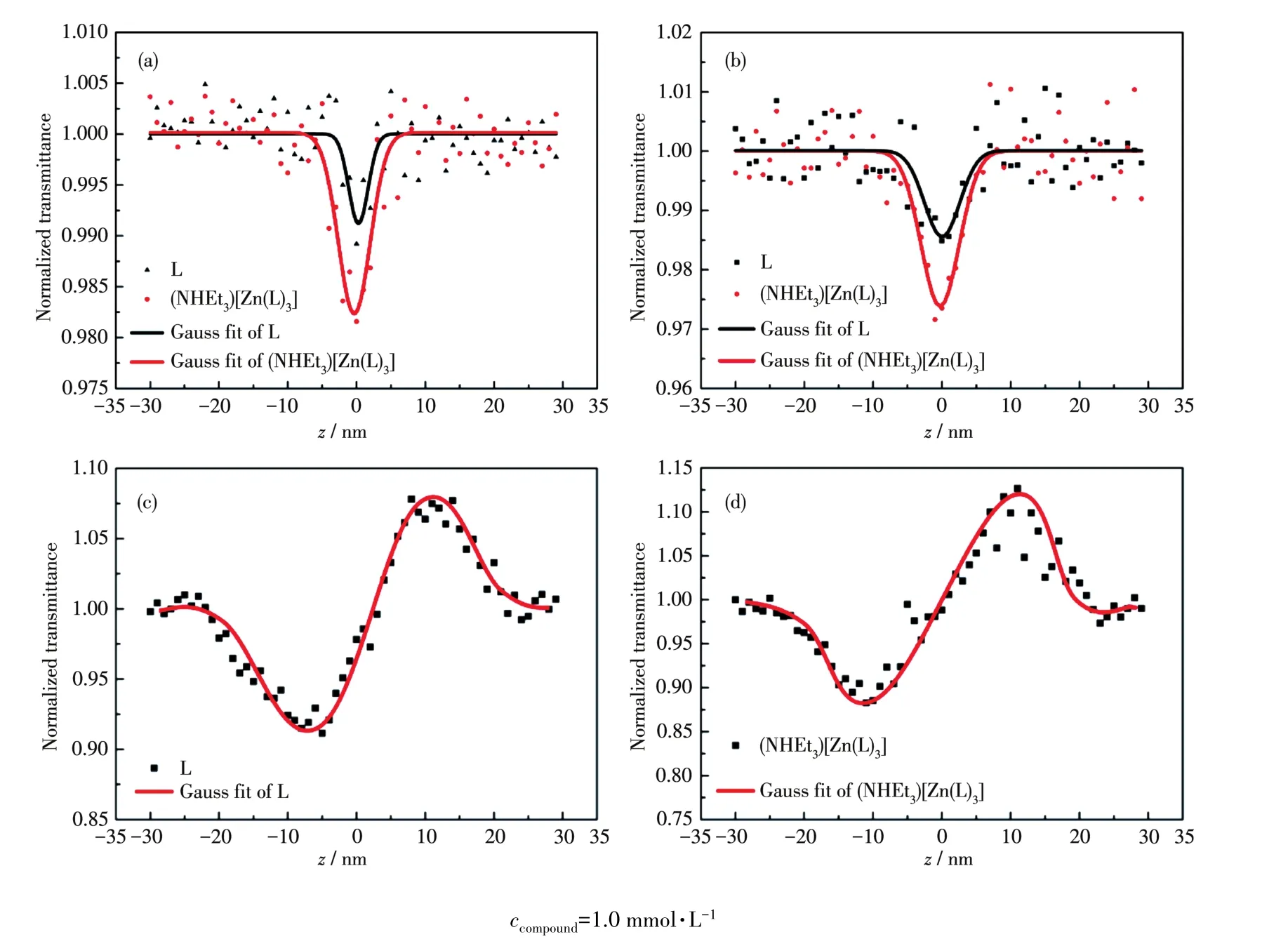
图10 在CH3CN溶液中,L和(NHEt3)[Zn(L)3]分别在850 nm(a)和1 350 nm(b)激发波长下的归一化开孔Z‑scan图;在CH3CN溶液中,L(c)和(NHEt3)[Zn(L)3](d)在850 nm激发波长下的归一化闭孔Z‑scan图Fig.10 Normalized open‑aperture Z‑scan transmittances of L and(NHEt3)[Zn(L)3]in CH3CN solution at 850 nm(a)and 1 350 nm(b),respectively;Normalized closed‑aperture Z‑scan transmittances of L(c)and(NHEt3)[Zn(L)3](d),respectively,in CH3CN solution at 850 nm excitation wavelength
通过非线性光学相关公式[28‑30]计算了配体和配合物在近红外区的三阶非线性极化率和多光子吸收效应参数,该配合物在850 nm附近有很强的双光子,在1 350 nm左右具有明显的三光子吸收效应。该配合物在近红外区表现出多光子吸收,明显是与它的分子内电子高度离域密切相关,其电子离域特性及电子转移机理已通过循环伏安和原位红外电化学法方法研究。
以上非线性光学性质研究显示:(1)通过增大化合物的共轭体系可以优化其非线性光学性质,而设计D‑A或D‑π‑A构型的分子或通过有机基团可以增强分子的共轭体系和非线性光学活性;(2)通过增强配合物分子内电子交流可以优化其非线性光学性质,而基于金属中心往往带正电荷的特性,设计带有负电性的有机配体,通过正负电荷之间的静电作用有利于增大配合物的稳定性,增强配合物分子内电子交流;(3)设计、合成多核金属配合物可以实现优化配合物的非线性光学性质。
3 结论
合成了一种电中性二茂铁双酮锌配合物(NHEt3)[Zn(L)3],通过单晶X射线衍射确定了配合物的结构,发现该配合物中二茂铁双酮以-1价烯醇式与锌金属配位,配合物外围存在一个质子化的三乙胺,以平衡整个分子的电荷。循环伏安和原位红外电化学研究表明,(NHEt3)[Zn(L)3]配合物经历了两步氧化和一步还原的准可逆氧化还原过程;结合理论计算发现β‑双酮二茂铁配合物分子内电子离域程度大,在近红外区表现出大三阶非线性极化率、双光子和三光子吸收效应,对设计、合成具有优良非线性光学性质的配合物有一定的指导意义。基于Fc与锌都具有低毒性和亲生物性的特点,结合本文性质研究结果,该二茂铁锌配合物在生物光动力治疗方面有进一步开发应用的价值。
Supporting information is available at http://www.wjhxxb.cn
猜你喜欢
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
铜仁学院学报(2018年6期)2018-07-05 09:47:34
固体火箭技术(2016年5期)2016-11-03 05:35:29
衡阳师范学院学报(2016年3期)2016-07-10 07:16:27
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
中学化学(2015年8期)2015-12-29 07:32:44
中国塑料(2015年5期)2015-10-14 00:59:47
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01 02:53:54
无机化学学报(2014年9期)2014-02-28 17:33:11

- 无机化学学报的其它文章
- Coexistence of Two Unique Cu(Ⅱ) Ions in Mononuclear Cu(Ⅱ)Complexes with Furanyl Substituted Triaryltriazoles
- Effect of Zn on Photocatalytic Activity of Block⁃Shaped Monoclinic WO3
- Synthesis,Structure,Luminescence,Photocatalytic and Magnetic Properties of a Neodymium Complex Constructed from Biphenyl⁃3,4′,5⁃tricarboxylic Acid
- Preparation and Nonlinear Absorption Properties of SiO2@CdTe@Au Composite Nanoparticles
- First⁃Principles Calculations on Electronic Structures and Optical Properties of g⁃C3N4Nanoribbons
- 基于(E)⁃N,N⁃二甲基⁃4⁃(2⁃(吡啶⁃4⁃基)乙烯基)苯胺的锌/镉有机-无机杂化金属卤化物的结构和发光性质