
Ru催化N2与H2反应合成氨的两态反应机理
2021-09-22 02:09吴晶晶王永成康娟霞
无机化学学报 2021年9期
吴晶晶 王永成 康娟霞
(西北师范大学化学化工学院,兰州 730070)
0 引 言
氨是工业生产和化肥合成不可或缺的原料,合成氨反应是20世纪最重要的科学进步成果之一[1‑2]。在过去几十年中,氨被广泛地应用于肥料行业,提高了粮食生产率,促进了世界人口的增长[3‑4]。而且近期研究发现,氨是一种重要的能量储存媒介和无碳能源载体[5‑6],这引起了研究者们对于合成氨的环保节能工艺研究的广泛关注[7‑9],因此,今后人类社会对氨的需求并不会减少。
合成氨反应的关键在于活化N2,N2键级为3,键能高达 941 kJ∙mol-1,因此,N2的活化需要极为苛刻的条件[10‑12]。在无催化剂条件下,H2活化 N2分子所需跨越的3个能垒,分别为523.84、240.58和274.05 kJ∙mol-1[13]。合成氨反应热力学行为虽然是放热反应,但反应能垒极高,常温常压下反应十分困难。催化剂在合成氨反应中能够活化N2,降低反应势垒,对于促进反应高效快速地发生起到极为关键的作用。因此,人们在开发高效能催化剂方面进行了大量的研究。Ru位于合成氨催化剂活性火山型曲线的顶端位置,表现出适中的N吸附能和较高的催化活性,催化性能甚至优于目前广泛使用的铁基催化剂[3,14‑15],具有高效能、高活性的特性。
Ru原子外层电子排布为4d75s1,d轨道电子未填满,存在自旋平行的单电子,既可以提供电子,也可以接受电子,具有较高的催化活性。但同时,反应中单电子可能受到自旋-轨道耦合作用而发生自旋翻转[16],使反应发生在2个或者多个势能面上,使得反应成为“两态反应”或“多态反应”[17‑21]。Ru催化合成氨反应的机理目前在理论研究方面尚有欠缺,特别是可能存在的“两态反应”现象更是常常被忽略。针对这一现象,采用密度泛函理论(DFT)[22‑24]计算方法,在计算单重态、三重态、五重态势能面的基础上,分析了Ru催化N2与H2反应合成氨过程中不同势能面之间的电子跃迁可能性,计算了两势能面最低能量交叉点[25](minimum energy crossing point,MECP)处的自旋-轨道耦合常数(HSOC)及系间窜越几率(PISC),并由此得到了反应的最低能量反应路径,得以深入理解Ru催化合成氨反应的微观机理。此外,为给理论研究和实际应用提供有用信息,利用Kozuch提出的能量跨度模型[26‑28]从理论层面计算Ru的转化频率(turnover frequency,TOF),确定了反应的决速过渡态(TDTS)和决速中间体(TDI),以期为合成氨催化剂的性能评估提供理论参考。
1 计算方法与理论背景
1.1 几何构型的优化
DFT已广泛应用于过渡金属化学的理论计算,采用(DFT)UB3LYP方法[29‑30],对 H、N 非金属元素采用 6‑311++G(3df,3pd)基组[31‑32],对过渡金属 Ru 采用SDD赝势基组。对反应体系的单重态、三重态、五重态势能面上的所有反应物、中间体、过渡态和产物的几何构型进行全参数优化和频率分析,得到了对应稳定结构的热力学物理量、零点矫正能和振动频率。确定各反应物、中间体和产物的能量为局部极小,各过渡态为鞍点,具有唯一振动虚频。过渡态到反应物和产物的基元步骤经内禀反应坐标(intrinsic reaction coordinate,IRC)[33]计算证实其可靠性。
本研究中主要计算工作运用Gaussian09程序[34]完成,反应物发生反应的活性位点预测、电荷分解分析、轨道相互作用图的绘制、静电势的计算及分析借助Multiwfn程序包[35]完成。反应中的键演变过程运用自然键轨道(natural bond orbital,NBO)方法分析。
1.2 自旋-轨道耦合常数及系间窜越几率计算
最低能量交叉点(MECP)的搜索运用Yoshizaw等的IRC单点垂直激发方法[36],借助Harvey的能量梯度优化法[37]得到。根据非绝热理论,不同自旋多重态在MECP处发生自旋态混合,在自旋-轨道耦合作用下,原准简并的势能面因消除简并发生分离,分离能为2HSOC,自旋-轨道耦合作用越强,分离能越大。自旋-轨道耦合能的计算采用Gamess程序[38‑39]完成。MECP的非绝热系间窜越几率(PISC)采用跃迁公式 Landau‑Zener公式(1~3)[40‑42]计算:
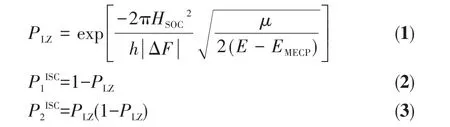
其中,PLZ为跃迁几率;h为普朗克常数;HSOC为不同自旋态之间自旋-轨道耦合矩阵元;ΔF为2个不同自旋态势能面之间的梯度差;μ为MECP的折合质量;E为反应物的相对能量;EMECP为MECP的相对能量。
1.3 能量跨度模型
催化TOF能够很好地反映催化剂的本征活性,是评价催化剂活性的重要指标[43]。本文中运用Kozuch提出的能量跨度模型和循环反应控制度[44‑45]的概念,从理论层面估算催化剂的TOF。ΔGr为总反应的Gibbs自由能差,对于ΔGr<0的反应,当确定了决速态(TDTS,TDI)后,就可得到TOF与能量跨度δE之间的近似关系式(4~5),δE即为催化反应过程的表观活化能。
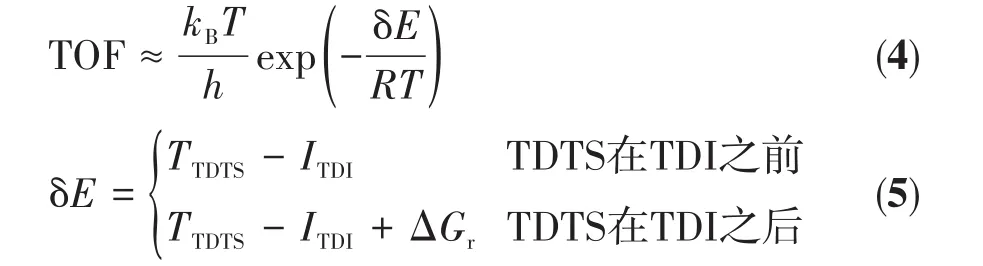
其中,TTDTS和ITDI分别为决速过渡态和决速中间体的Gibbs自由能,kB为玻尔兹曼常数,T为温度,R是理想气体常数。
Kozuch等参照Compbell对循环反应速率控制度的表示,定义了TOF控制度XTOF的概念,某一态(过渡态i或者中间体j)的XTOF值越大,该态对TOF的影响越大。用Ti和Ij表示第i过渡态和第j中间体的Gibbs自由能,ΔGij表示为态 i和态 j的 Gibbs自由能差。用能量准确表示XTOF则可以写成:

各态的XTOF需符合公式:
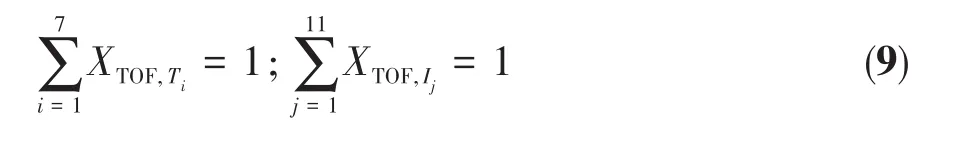
2 结果与讨论
Ru原子外层电子组态为4d75s1,基态为五重态5Ru,受到激发时,5s轨道上的单电子跃迁到4d轨道,第一激发态为三重态3Ru,第二激发态为单重态1Ru。为考察Ru原子对N2与H2的催化循环反应,分别计算了Ru在单重态、三重态、五重态下催化N2与H2反应的势能面,在多势能面上对反应进行了综合分析讨论。
2.1 几何构型优化及构型参数
对Ru催化N2与H2反应生成氨过程中各基元反应的中间体及过渡态进行全几何构型优化,各驻点的结构及参数如图1所示(仅列出三重态,单重态、五重态各驻点及参数列在图S1、S2中,Supporting information),能量列在表S1中。
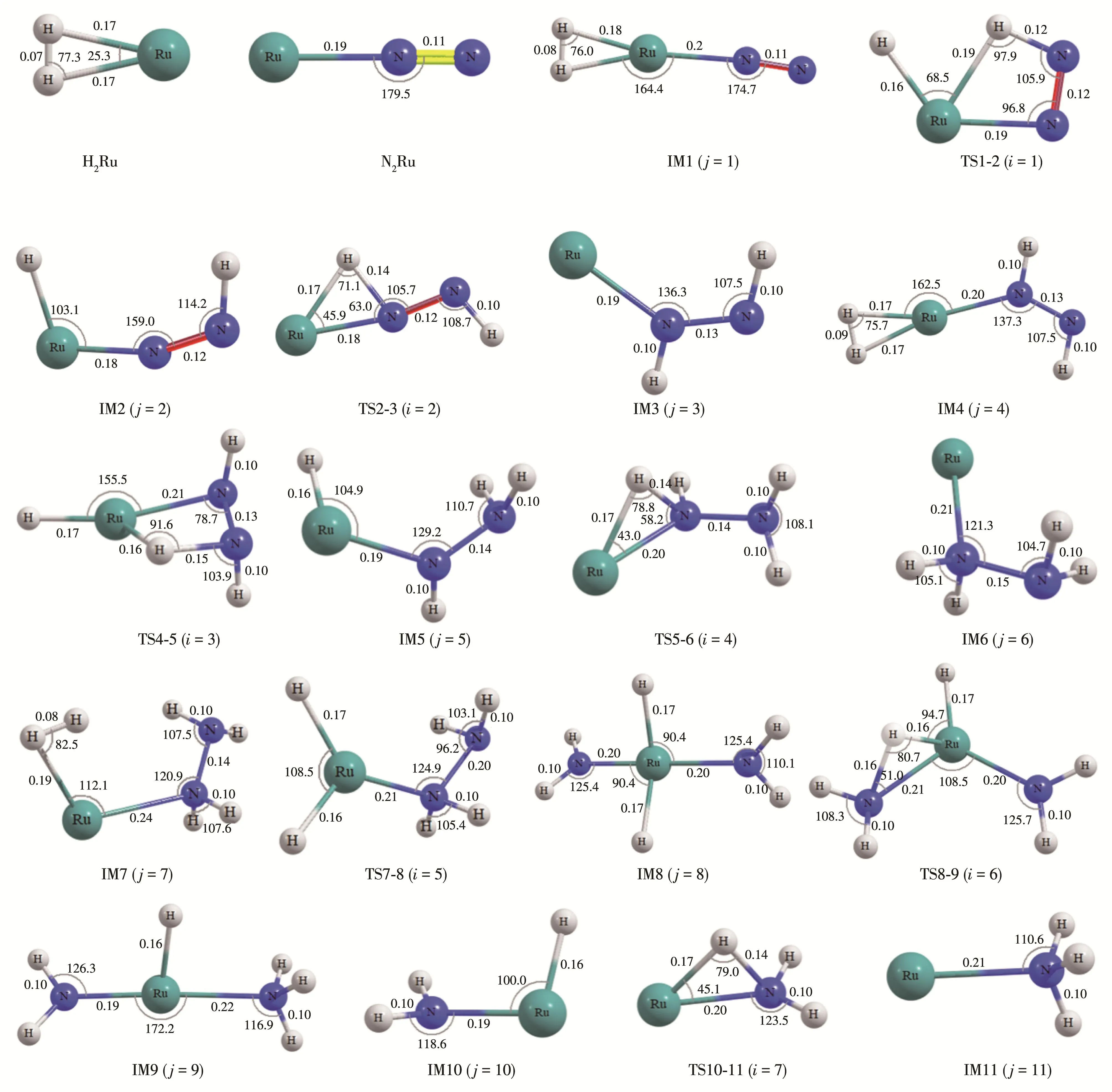
图1 B3LYP/6‑311++g(3df,3pd)∪sdd水平下三重态势能面各驻点的几何构型(键长为nm,键角为°)Fig.1 Optimized geometries for stationary points on triplet potential energy surfaces at the B3LYP/6‑311++g(3df,3pd)∪sdd level(Distances in nanometer and angles in degrees)
2.2 反应位点预测
经计算,基态五重态具有最低能量,反应以五重态进入反应通道。Ru同时与N2、H2相互作用时,存在2种可能的反应方式:(1)Ru与H2形成Ru‑H2后再与 N2反应。(2)与(1)相反,Ru 与N2形成 Ru‑N2后再与H2反应。
分析Ru、N2、H2的前线分子轨道(FMO)(图2),Ru的LUMO轨道、SOMO轨道分别与N2、H2的HOMO、LUMO轨道对称性匹配。5Ru的LUMO轨道与N2、H2的HOMO轨道能差均十分接近,分别为1 084.33和1 066.74 kJ·mol-1。
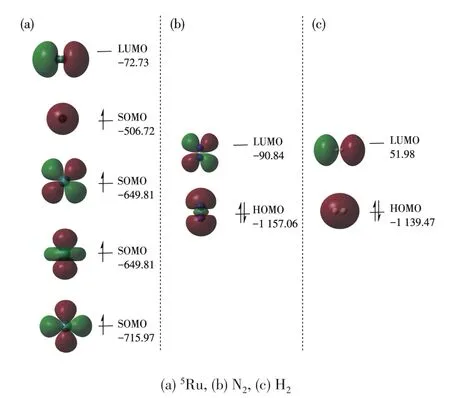
图2 5Ru、N2、H2的FMOs(能量单位:kJ·mol-1)Fig.2 FMOs of5Ru,N2and H2(Energy unit:kJ·mol-1)
运用 Multiwfn 程序中的静电势[46‑47]计算、预测反应活性位点,图3显示了5Ru、N2、H2、N25Ru及H25Ru的表面静电势极值点。5Ru的静电势极值点均为正值(图3c),静电势极大值 Vs,max=35.06 kJ∙mol-1,正电位与负电位相互吸引,静电势极大值处容易吸引亲核试剂。H2有环键轴的等值负电位,静电势极小值Vs,min=-11.17 kJ·mol-1,H 只有一个电子,涉及共价键,通常很少形成线性结构。Ru原子将会以垂直H2键轴方向进攻,静电势极大值处与H2的环键轴负电位相互作用,与2个H原子分别成键,形成H2Ru(图3d),结合能 ΔE=20.31 kJ·mol-1。N2在 N 端有 2 处负电位极小值,Vs,min=-34.94 kJ·mol-1,N的负电性比H更强,Ru从N2的N端位进攻,在非共价作用力下形成线性结构,结构见图3e,结合能ΔE=13.32 kJ·mol-1,低于形成H2Ru时的结合能。
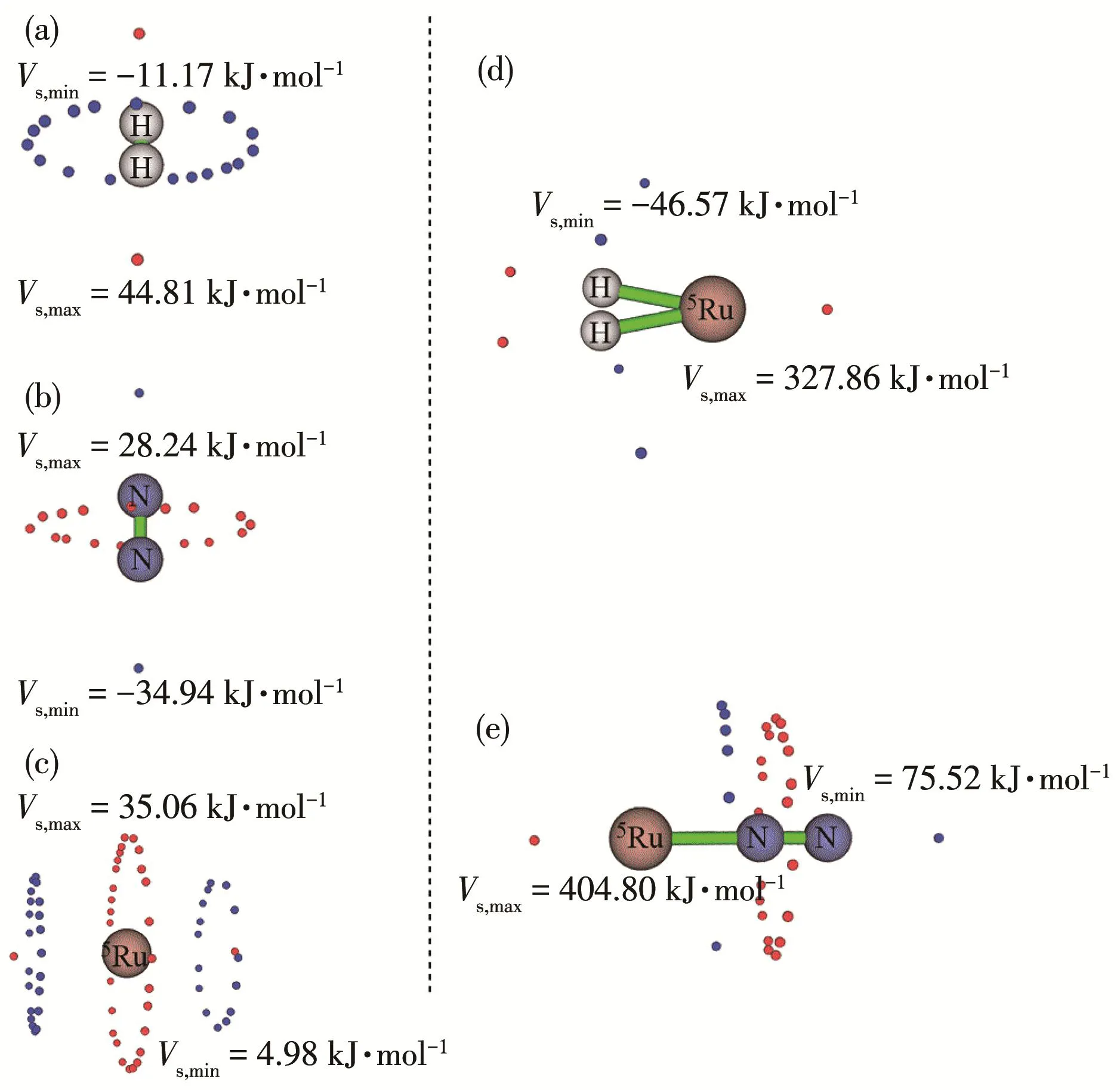
图3 (a)H2、(b)N2、(c)Ru、(d)H25Ru和(e)N25Ru的表面静电势极值点分布图(红色为极大值,蓝色为极小值)Fig.3 Extreme value points of surface electrostatic potential of(a)H2,(b)N2,(c)Ru,(d)H25Ru,and(e)N25Ru(red is the maximum and blue is the minimum)
由于N的电负性,引起Ru—N—N的电子密度由Ru向N偏移,Ru端电子密度减小,极化增强。N2Ru的Ru端具有较高的正电位,静电势极大值Vs,max=404.80 kJ·mol-1,H2Ru 中 Ru 端 的 静 电 势 为327.86 kJ·mol-1,相比较而言,N2Ru的静电势极大值处静电势能更强。静电势越大,Ru端σ‑穴更易与负电位相互作用,更容易与亲核试剂发生反应。从静电势的角度出发,可以认为N2Ru更为稳定,5Ru优先与N2结合。
2.3 电荷分解分析
基态5Ru能量比3Ru能量低 74.04 kJ·mol-1,比1Ru能量低236.30 kJ·mol-1,反应沿五重态势能面进入反应通道,Ru优先与N2相互作用。形成N2Ru时,单重态势能面能量降低了170.46 kJ·mol-1,三重态能量降低了105.62 kJ·mol-1,五重态能量升高了13.32 kJ·mol-1,不同自旋态势能面上N2Ru的形成表现出差异性。以N23Ru、N25Ru为例,运用Multiwfn程序对N2Ru复合物进行电荷分解分析(charge decom‑position analysis,CDA)[48‑49]。
由于体系存在未配对的电子,CDA分析中采用UB3LYP非限制性开壳层计算方法,定义Ru为片段1,N2为片段2,对三重态/五重态N2Ru的α及β轨道分别进行研究,轨道相互作用如图S3所示,三重态/五重态N2Ru的HOMO、LUMO能量及HOMO‑LUMO能差见表S2和图4。
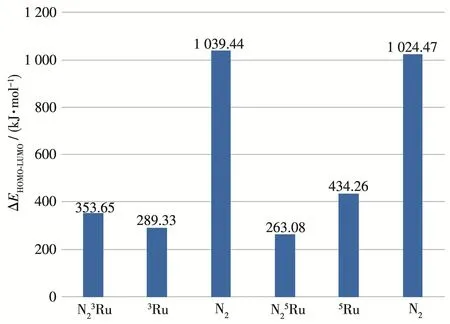
图4 三重态/五重态N2Ru α轨道的HOMO‑LUMO能差ΔEFig.4 HOMO‑LUMO gap ΔE of α orbitals of triplet/quintet N2Ru
分析显示,N23Ru α轨道的HOMO‑LUMO能差为353.65 kJ·mol-1,比3Ru 增 大 了 64.32 kJ·mol-1。的HOMO轨道和LUMO轨道主要由3Ru原子轨道贡献构成。3Ru的LUMO+1轨道参与形成LUMO轨道,造成了N23Ru的LUMO轨道能级抬高,HOMO‑LUMO能差变大,3Ru向N2净转移电子数0.004 3(图5)。β轨道中,3Ru的HOMO‑LUMO能差为186.41 kJ·mol-1,N23Ru的 HOMO‑LUMO 能 差 为177.48 kJ·mol-1,能差变化不大。N25Ru α 轨道的HOMO‑LUMO能差为263.08 kJ·mol-1,较Ru减小了171.18 kJ·mol-1,β轨道仅增大了28.62 kJ·mol-1。
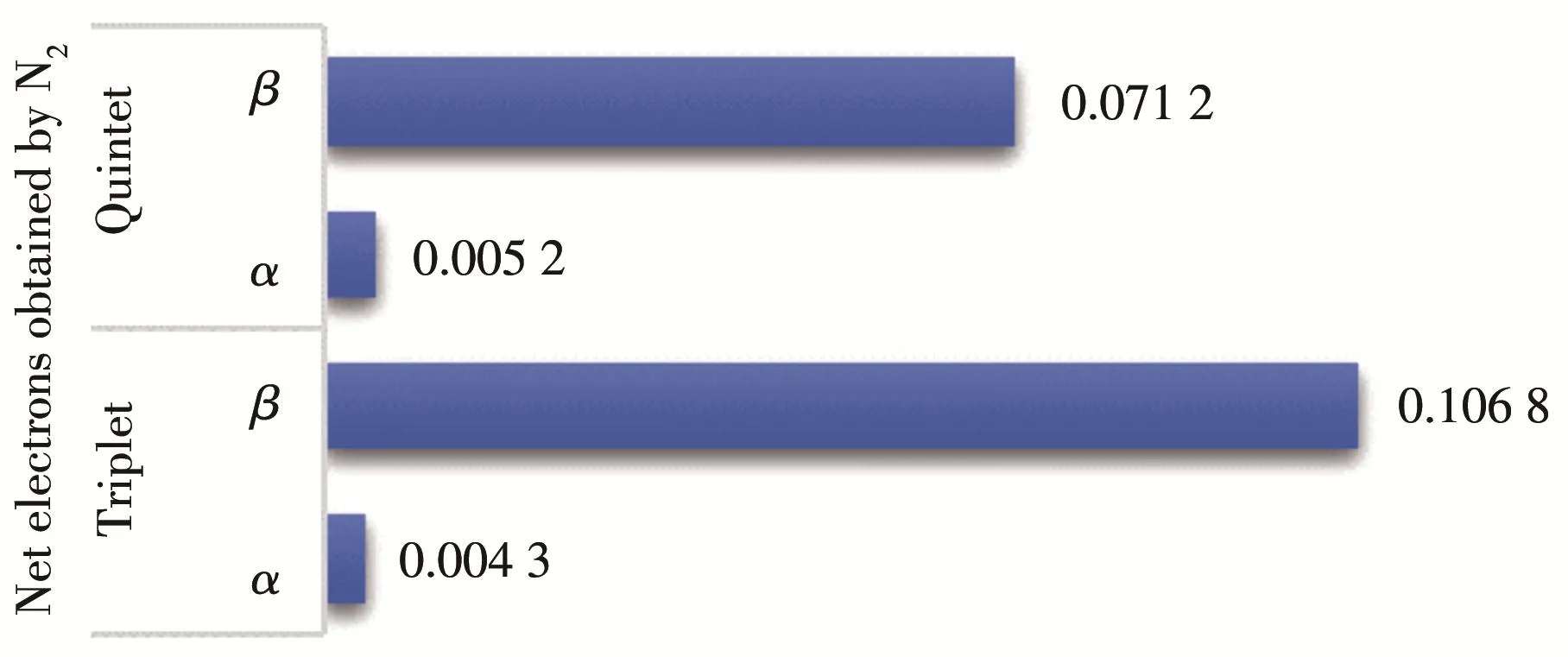
图5 三重态/五重态N2Ru复合物Ru向N2净转移电子数Fig.5 Net electrons transferred from Ru to N2on the triplet/quintet N2Ru
总体来讲,N23Ru复合物HOMO‑LUMO能差在3Ru原子及N2分子轨道线性组合过程中显著增大,N25Ru的HOMO‑LUMO能差明显减小,因此,三重态复合物更难发生电子的跃迁,N23Ru较N25Ru更为稳定。另一方面,在形成N2Ru时,三重态α轨道及β轨道总的净转移电子数为0.111 1,而五重态为0.076 4,三重态净转移电子数更多,更有利于N2的解离,促进反应进行。
2.4 势能面交叉
N23Ru比N25Ru更稳定,能量更低,体系在N2Ru前形成势能面交叉点CP1(图6),如果能够在该处发生有效系间窜越,反应体系将由五重态跃迁到三重态势能面上进行。
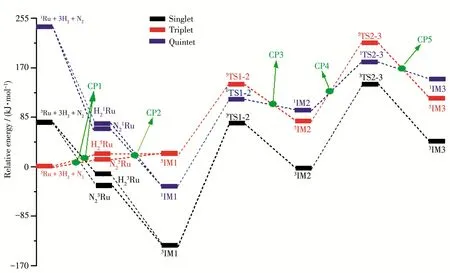
图6 B3LYP/6‑311++g(3df,3dp)∪sdd水平下Ru催化N2与H2反应的反应势能面图Fig.6 Potential energy surface diagram of reaction between N2and H2catalyzed by Ru at the B3LYP/6‑311++g(3df,3dp)∪ sdd level
N2Ru中Ru端基结合H2,同时使H—H解离形成IM1,接下来是连续两步的H向N转移过程,H转移基元反应的过渡态3TS1‑2及3TS2‑3的虚频分别为933.39和1 055.76i cm-1,振动方向均为H自Ru向N转移的振动方向。能垒分别为207.15、140.99 kJ·mol-1,远低于无催化剂的合成氨加氢反应能垒(523.84 kJ·mol-1)[13],极大地促进了反应进行。3IM3的NBO分析结果为:σ(N2—N3)1=0.732 9(sp1.56)N2+0.680 4(sp2.35)N3,π(N2—N3)2=0.769 4(sp)N2+0.638 7(sp)N3,说明3IM3已形成N—H键,原N≡N叁键中的一个π键断裂,形成RuN2H2。
图7为RuN2H2与H2反应的势能面图,由图可见,加H2反应仍然在三重态势能面上完成。插H2过程与上步反应类似,H2被Ru的正电位吸引形成复合物,同时解离H—H键。接着连续两步发生由Ru到N的H转移,形成IM6[RuN2H4],3TS4‑5和3TS5‑6分别是H转移的过渡态,H原子转移分别需要跨越113.94和123.66 kJ·mol-1的能垒,相比较无催化剂合成氨反应能垒(240.58 kJ·mol-1)而言,能垒也有大幅度降低。
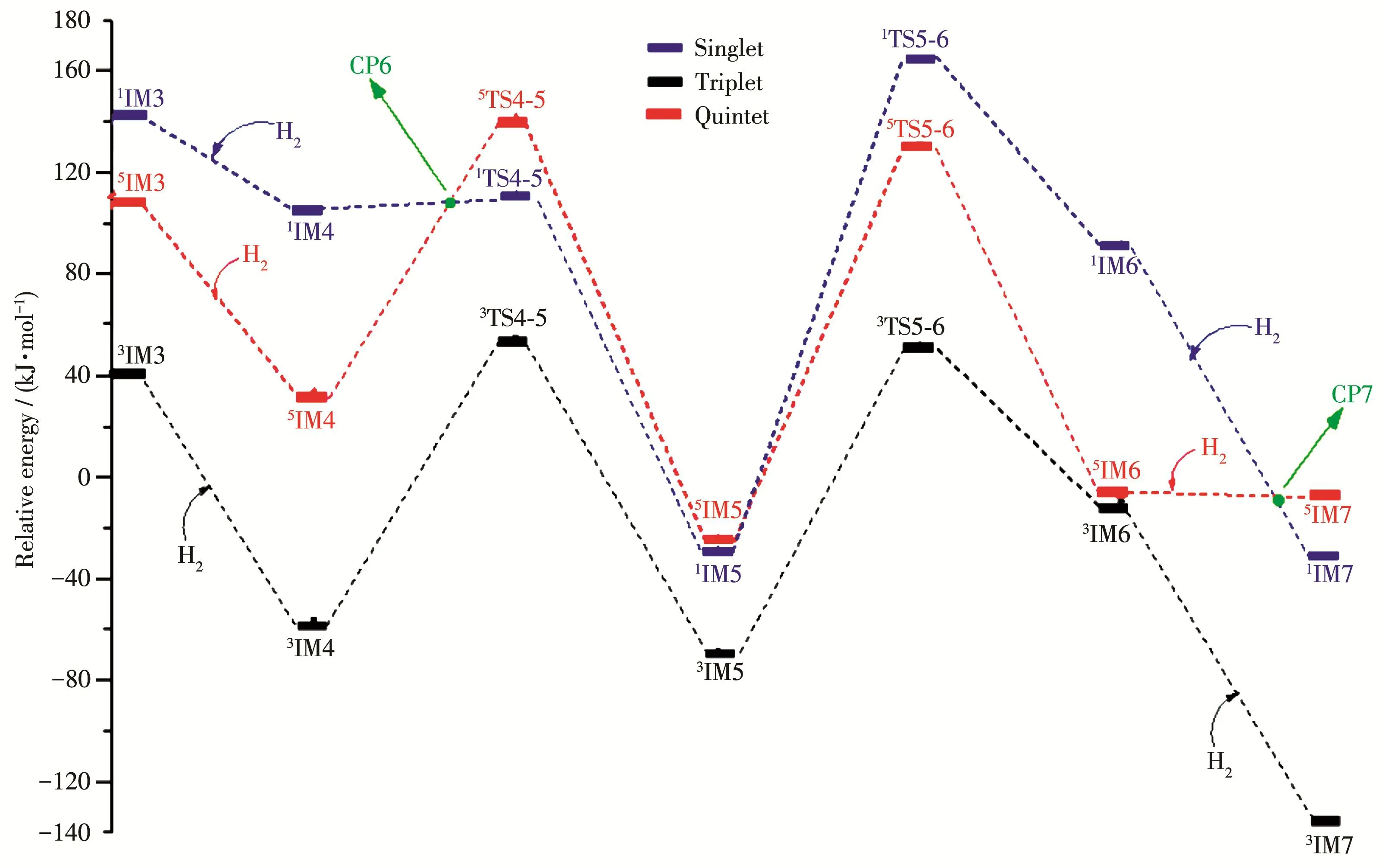
图7 B3LYP/6‑311++g(3df,3dp)∪sdd水平下RuN2H2与H2反应的势能面图Fig.7 Potential energy surface diagram of the reaction between RuN2H2and H2at the B3LYP/6‑311++g(3df,3dp)∪ sdd level
如图8所示,Ru上插入第3个H2形成3IM7((H2)RuN2H4)后,经能 垒仅为 7.88 kJ·mol-1的过渡 态TS7‑8后,N—N键断裂,体系能量大幅度下降。继续发生H转移过程,3TS8‑9有唯一虚频1 136.21i cm-1,能垒为 85.85 kJ·mol-1,过渡振动矢量为 H 自Ru向N的方向。从3IM9((H2N)HRuNH3)中脱离一分子NH3后形成3IM10(HRuNH2),再经过渡态3TS10‑11将Ru上的H转移到NH2上形成IM11(3RuNH3)。第2个NH3分子从Ru原子上解离,自旋态由三重态变为五重态,即3RuNH3→5Ru+1NH3,说明脱NH3势能面上存在三重态与五重态势能面交叉点CP9。整个反应为放热反应(ΔE=-177.54 kJ·mol-1)。
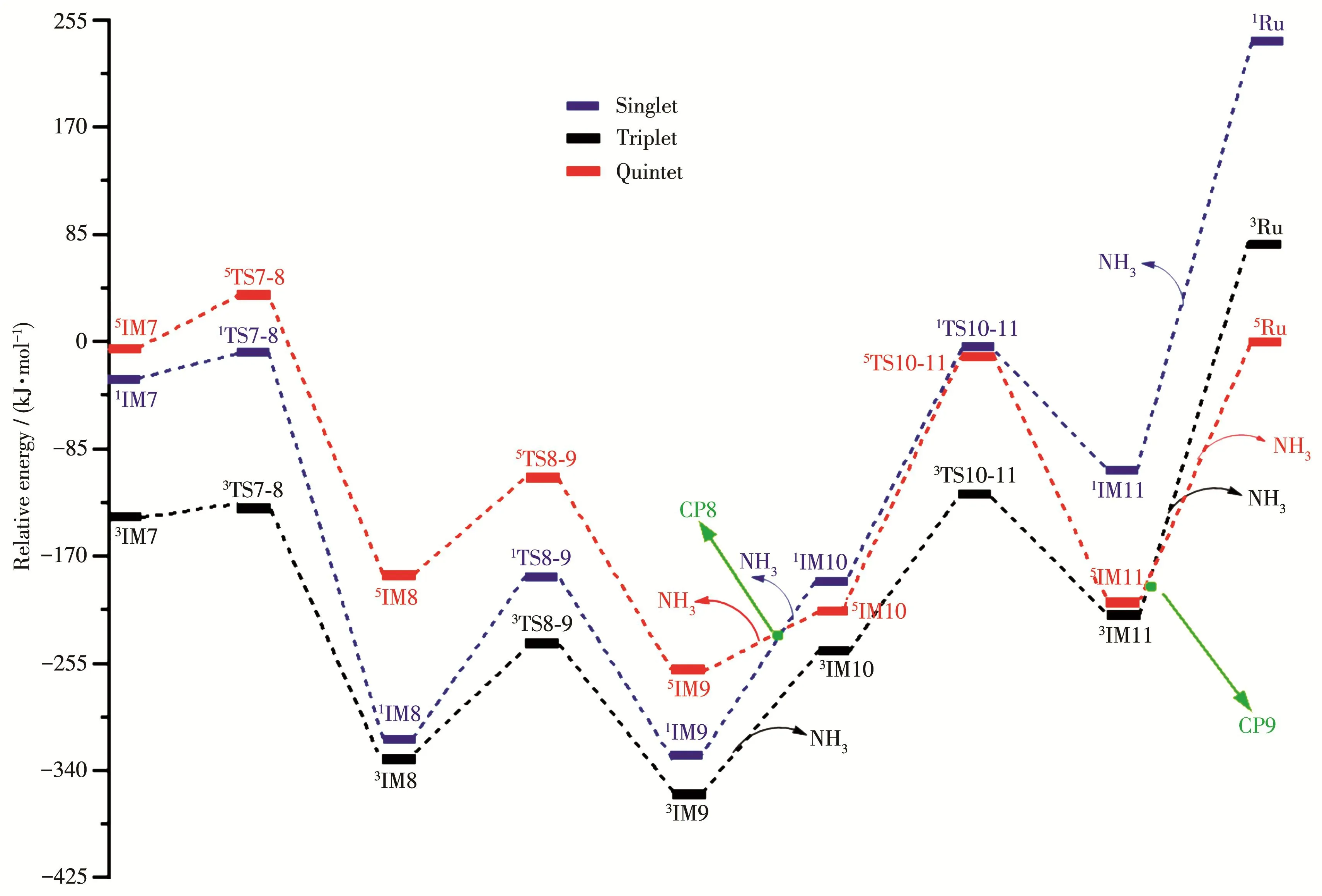
图8 B3LYP/6‑311++g(3df,3dp)∪sdd水平下RuN2H4与H2反应的反应势能面图Fig.8 Potential energy surface diagram of the reaction between RuN2H4and H2at the B3LYP/6‑311++g(3df,3dp)∪ sdd level
2.5 最低能量交叉点
从图6~8中可以看出,在整个催化过程中,Ru体系催化合成氨反应主要在三重态势能面上完成,Ru以基态五重态(5Ru)进入反应通道,又以五重态(5Ru)离开反应通道,反应在单重态/五重态、三重态/五重态势能面存在多个势能面交叉点,但仅有反应进、出口附近存在的CP1、CP9两个交叉点对反应可能存在的低能反应路径有贡献,因此,我们仅对三重态/五重态势能面交叉点CP1及CP9进行分析。
在三重态、五重态上分别对N2Ru和Ru‑NH3做构型能量相关扫描,得到初步交叉点CP1和CP9,在此基础上运用Crossing2004程序包[35]优化得到相应的最低能量交叉点MECP1及MECP9结构(图9),以及势能梯度等信息。

图9 三重态/五重态(a)MECP1及(b)MECP9结构参数Fig.9 Structural parameters of triplet/quintet(a)MECP1 and(b)MECP9
有效的自旋-轨道耦合是自旋禁阻反应发生系间窜越的主要机制,自旋-轨道耦合作用越强(即Hsoc值越大),MECP处原简并的能量分离程度越大,即上、下能差(ΔE=2Hsoc)越大,上、下绝热势能面之间的非绝热跃迁几率越小,不同自旋态势能面间的系间窜越几率就越大。运用Gamess程序计算得到MECP1处的自旋-轨道耦合常数Hsoc=508.34 cm-1,运用系间窜越公式估算得到MECP1处的单程系间窜越几率P1>ISC=0.25,双程系间窜越几率P2ISC=0.85,MECP9处Hsoc=269.21 cm-1、P1ISC=0.16、P2ISC=0.27。较大的系间窜越几率说明在MECP处的非绝热窜越速率较大,体系在MECP处容易通过系间窜越到低势能面上,确保反应沿低能反应路径上进行。
图10即为MECP1及MECP9的FMO能级图。以MECP1为例,体系在MECP处发生系间窜越时,受到自旋磁矩与轨道磁矩的相互作用,原来的电子自旋态发生改变,Ψa是三重态的最低空轨道,也是五重态的最高单占轨道,主要由Ru的s轨道组成;Ψb、Ψc、Ψd能级相近,主要由Ru的dz2、dxz、dyz组成,当体系沿反应路径到MECP1处时,借助自旋-轨道耦合作用,使五重态上Ψa轨道中自旋向上的α电子翻转为自旋向下的β电子,并跃迁到Ru的d轨道上,与原Ψd上的α电子自旋配对,Ψa成为空轨道,从而完成五重态向三重态的系间窜越过程。
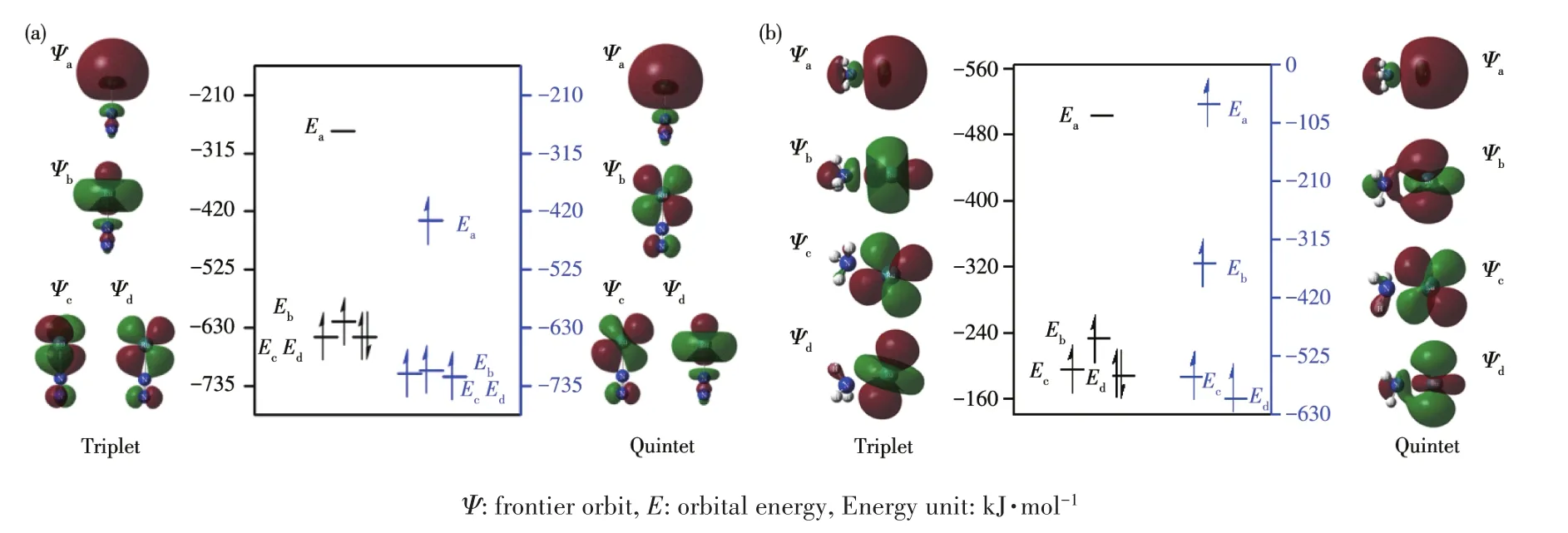
图10 MECP1(a)和MECP9(b)的FMO能级图Fig.10 FMO interaction analysis for MECP1(a)and MECP9(b)
运用Multiwfn软件计算MECP处的自旋密度(spin density)和自旋布居(spin population)以分析MECP处的单电子分布情况[50]。从表1及表2可以看到MECP1及MECP9中三重态的2个单电子主要分布在Ru原子上,分别为2.13及2.04;五重态中,Ru在MECP1及MECP9的总自旋布居分别为4.09和3.98,N、H原子的总自旋布居极小,表明4个单电子也主要分布在Ru的原子轨道上。这一情况同样体现在电子自旋密度(图11)中,单电子主要集中分布在Ru原子上,N、H原子上几乎不存在单电子。MECP1及MECP9处电子自旋密度形态十分接近。电子的自旋翻转主要发生在体系的Ru原子上,此结果符合自旋-轨道耦合作用的重原子效应理论。

表1 MECP1的自旋布居Table 1 Spin population for MECP1
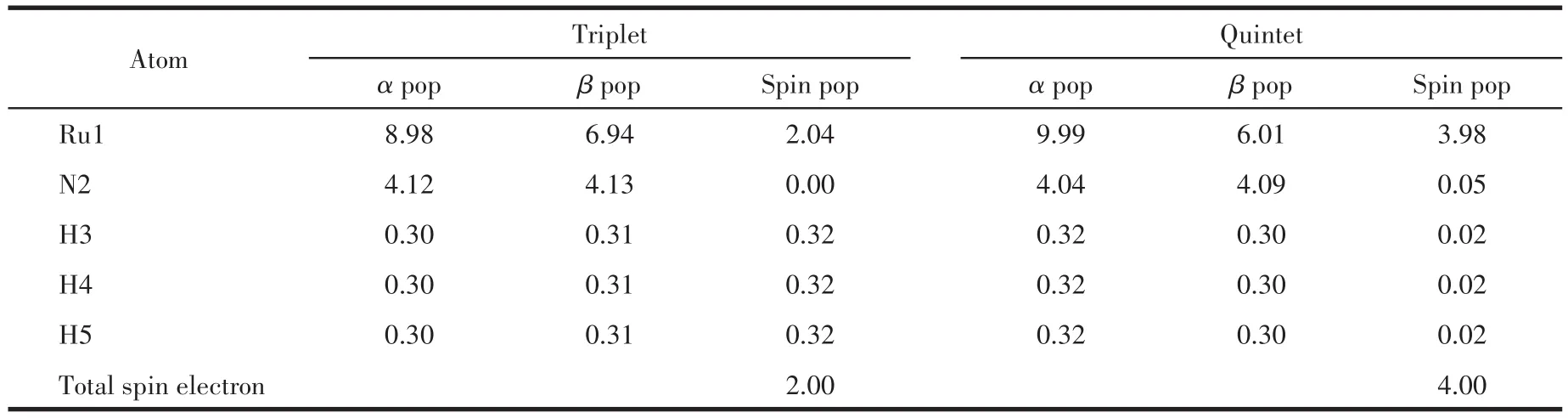
表2 MECP9的自旋布居Table 2 Spin population for MECP9
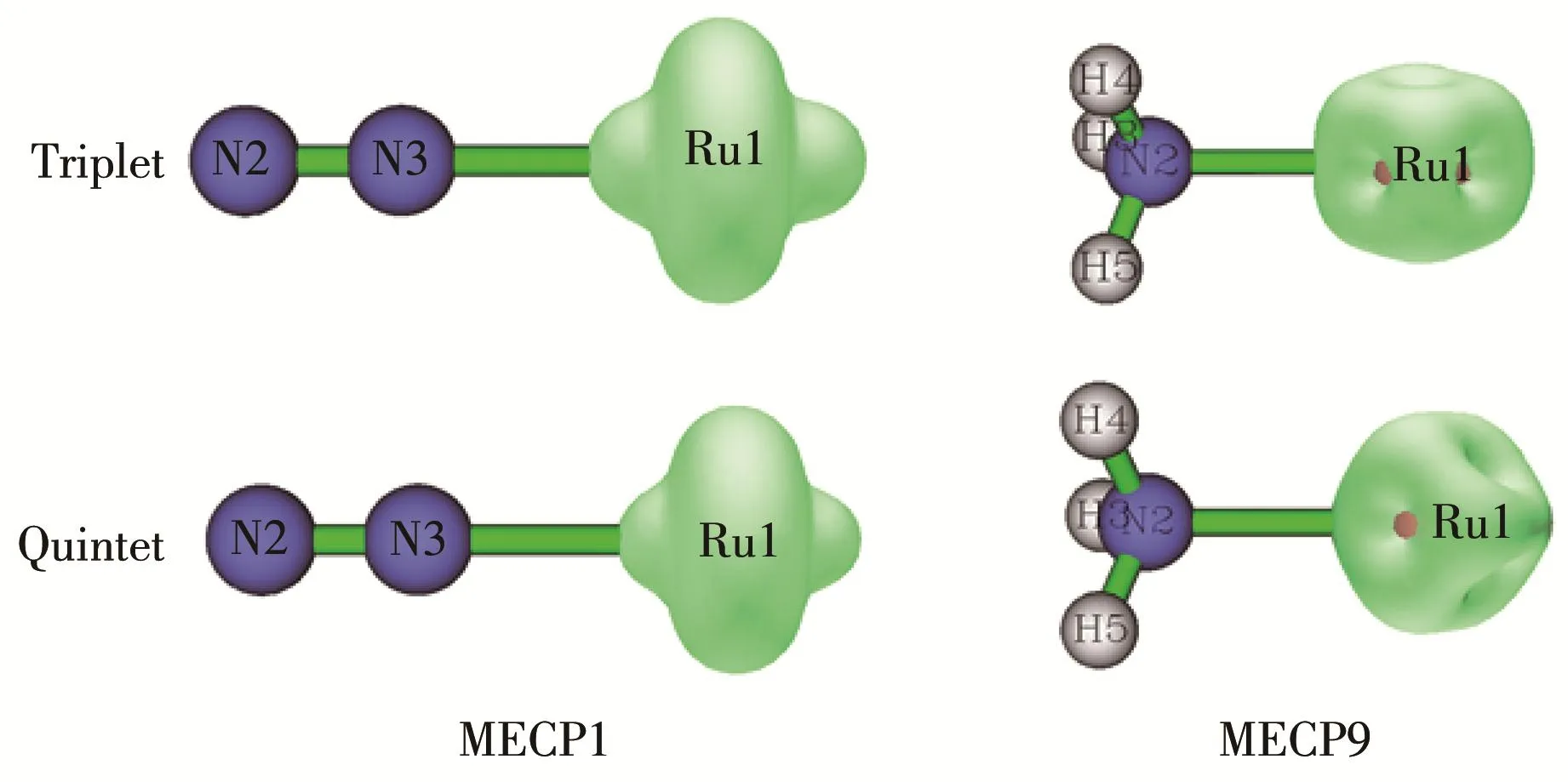
图11 MECP1和MECP9的电子自旋密度图Fig.11 Electron spin density of MECP1 and MECP9
2.6 非绝热反应的TOF计算
TOF值衡量的是一个催化剂催化反应的速率,表示的是催化剂的本征活性,能够很好地反应出催化剂的催化性能。虽然在多相催化体系中很难准确计算TOF值,但是对于同一个模型反应,TOF可用于比较不同催化剂的本征催化活性,可用于评估催化剂的发展潜力。Kozuch提出的能量跨度模型为在理论层面计算TOF值[43‑45]以横向比较催化剂的催化性能提供了一个可利用的工具。
我们运用能量跨度模型对Ru催化N2与H2循环反应的TOF值进行理论计算。图12为反应的最低能量反应路径的Gibbs势能图,整个催化循环反应是正向反应,反应的相对Gibbs自由能为ΔGr=-47.73 kJ·mol-1。需跨越的能垒最高的过渡态为3TS1‑2,能垒为207.15 kJ·mol-1,能量最高的过渡态为3TS2‑3,相对能量为 139.70 kJ·mol-1(图 6)。虽然是正向反应,但过高的能垒限制了反应速率,在常温常压条件下反应几乎不可能发生。
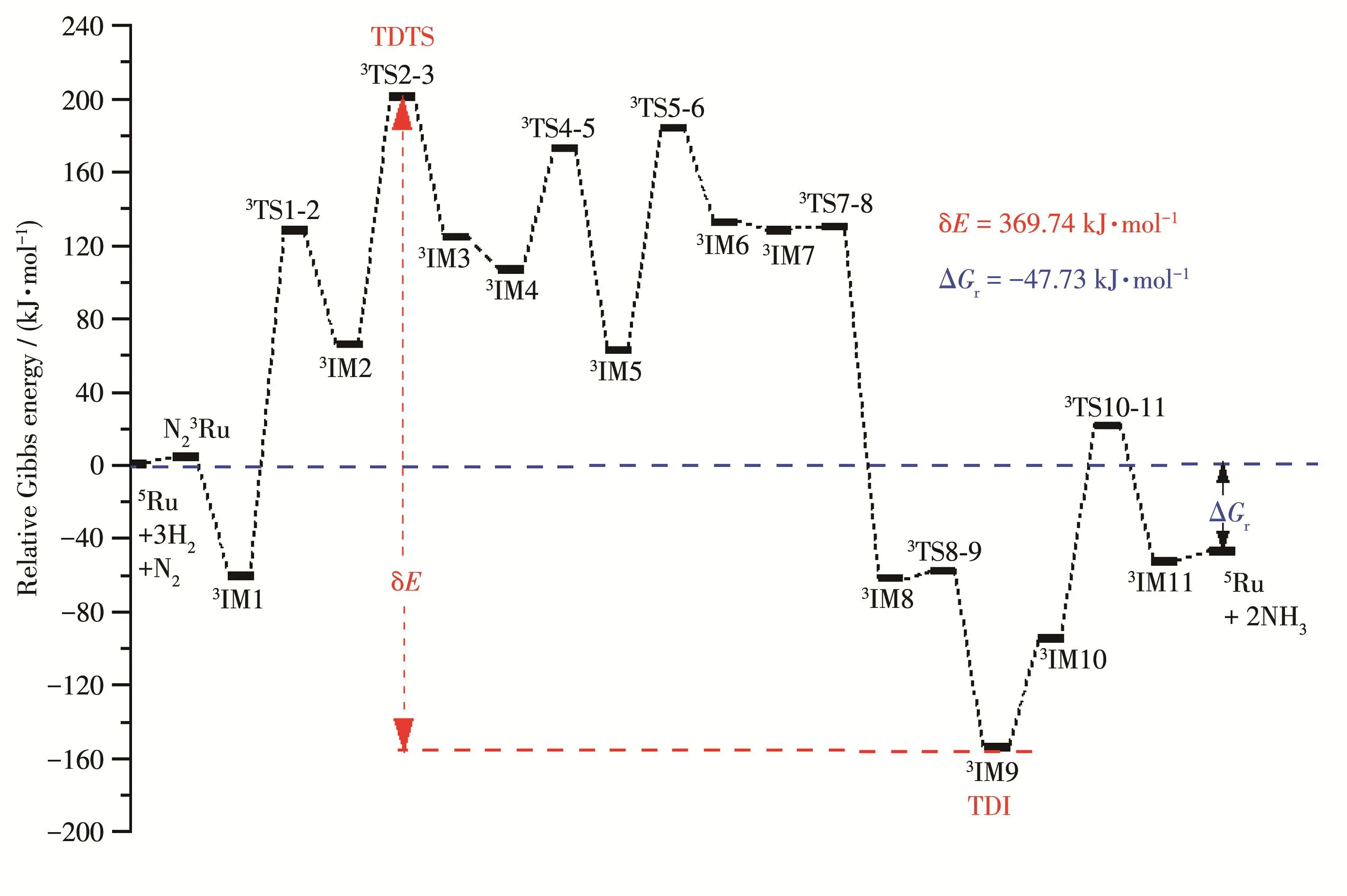
图12 Ru催化N2+3H2→2NH3循环反应的ΔGr图Fig.12 ΔGrdiagram of N2+3H2→ 2NH3cyclic reaction catalyzed by Ru
能量跨度模型认为,在一个催化循环中,各中间体和过渡态的Gibbs自由能对总速率都有影响,但对反应总速率起到决定性作用的是催化循环过程中Gibbs自由能最高的TDTS和最低的TDI,且TDTS与TDI可不相邻。TDTS与TDI态间自由能差值(能量跨度δE)决定了整个催化循环反应速率的大小。
3TS2‑3与3IM9分别是反应中Gibbs能量最高和最低的过渡态和中间体,各中间体和过渡态对反应的控制度可以用XTOF来表示,部分态的XTOF如表3所示,经计算,在298 K下,3TS2‑3与3IM9同样分别是反应的 TDTS(XTOF,TS2‑3=0.98)和 TDI(XTOF,IM9=1.00),其他中间体和过渡态的XTOF值均近似等于0,可以忽略不计。

表3 最低能量反应路径的各中间体和过渡态对反应的XTOFTable 3 XTOFof each intermediate and transition of minimum energy path against the reaction
由于ΔGr<0、TDTS位于TDI之前,经由公式得出反应的能量跨度δE和TOF值:298 K下,δE=369.74 kJ·mol-1,TOF=6.84×10-36s-1。TOF值较小,表明该体系在理想状态下反应速率较小,符合合成氨工业较难发生的反应特征,但与相同状态下FeO催化合成氨反应相比,Ru催化合成氨反应的TOF值远大于FeO的TOF值1.24×10-60s-1[51],Ru不失为一种提高合成氨反应效率,取代铁基催化剂的有效催化剂。
3 结论
采用DFT在 B3LYP/6‑311++g(3df,3pd ∪ sdd)水平上,对Ru催化N2+3H2→2NH3的循环催化反应进行了理论研究,结果表明:
(1)N2优先于H2与Ru结合,在Ru位点上加H2的同时,H—H键解离。H转移过程只发生在Ru与N之间,与FeO相比,H转移过程更为简单;
(2)Ru催化N2与H2循环反应是两态反应,在绝热状态下,Ru以基态五重态进入反应通道,经由入口交叉点处发生有效系间窜越,反应主要在三重态势能面上进行,最后在反应出口交叉点处仍经系间窜越,以基态五重态离开反应通道,反应沿低能路径进行,反应的最低能量路径:
5Ru→N23Ru →3IM1→3TS1‑2 →3IM2→3TS2‑3→3IM3→3IM4→3TS4‑5 →3IM5→3TS5‑6→3IM6→3IM7→3TS7‑8→3IM8→3TS8‑9→3IM9→3IM10→3TS10‑11 →3IM11→5Ru
(3)该反应为正向放热反应,反应中能量最高的过渡态为TS2‑3,较高的能垒使得升高温度有利于反应发生;
(4)运用Kozuch能量跨度模型计算298 K下Ru催化N2+3H2→ 2NH3循环反应的能量跨度δE(δE=369.74 kJ·mol-1)和 催 化 转 化 频 率(TOF=6.84×10-36s-1),反应的决速过渡态为3TS2‑3,决速中间体为3IM9。Ru的催化频率TOF值优于FeO催化剂,可作为提高合成氨反应效率的有效催化剂。
Supporting information is available at http://www.wjhxxb.cn
猜你喜欢
中国化肥信息(2022年3期)2023-01-05
中国化肥信息(2022年4期)2023-01-02
北京航空航天大学学报(2022年5期)2022-06-06
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
燃料化学学报(2021年5期)2021-06-02
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
山东工业技术(2016年15期)2016-12-01
科技视界(2016年24期)2016-10-11
