
Co⁃Co9S8/N掺杂科琴黑复合材料的制备及其氧还原反应催化性能
2021-09-22 02:10李福枝孙翱魁贺全国
无机化学学报 2021年9期
李福枝 陈 真 张 丹 孙翱魁 石 璞*, 贺全国
(1湖南工业大学包装与材料学院,株洲 412008)
(2湖南工业大学生命科学与化学学院,株洲 412008)
金属-空气电池(metal‑air battery)作为新一代燃料电池,不仅具有较高的理论能量密度,而且在成本、环保性和安全性方面极具优势,因此受到了研究者们的广泛关注。其中,铝-空气电池理论能量密度可高达600 Wh·kg-1,在新能源电动汽车领域的应用前景广阔[1‑2]。然而,这种电池的主要挑战在于如何提高氧还原反应(oxygen reduction reaction,ORR)速率,从而提高电池的能量效率。目前,研究者们主要利用催化剂来提高ORR速率。Pt/C催化剂被证实以4电子机制催化ORR,被普遍用作评价ORR催化剂的参照物。然而,其高价格和稀缺性限制了其应用。因此,研制出非铂基、低成本、高性能的ORR催化剂是非常重要的。过渡金属硫化物作为新型的ORR催化剂,具有取代Pt基催化剂的广阔前景[3‑4]。在众多过渡金属硫化物材料中,多形貌、多物相种类、资源丰富的硫化钴最引人关注。然而,活性位点单一、电子导电性低的缺点限制了它的应用。将硫化钴与高导电碳复合是解决上述问题的方法之一。例如,Han等[5]通过在惰性气体保护下高温热解聚多巴胺包覆的碱式碳酸钴纳米线和硫磺粉混合物,制备了N、S共掺硫化钴纳米线/石墨碳的核壳结构复合物(CoS NWs@NSC),纳米线叠加堆积形成三维导电网络,加快了电子在材料表面的转移,使得其ORR催化活性大大增强,可与商业Pt/C相当。Ding等[6]利用生物模板法制备了介孔N掺杂碳复合硫化钴纳米片(CoS/NC),表现出比商业Pt/C更好的ORR催化活性和稳定性;将其组装成锌-空气电池时,其放电电压平台可达到1.44 V。
近年来,单金属催化剂因具有纳米结构及可以达到尺寸更小的单原子态而具有高效的原子利用率和催化能力,在催化领域引起了广泛的关注。有研究者发现通过多级锚定策略,可实现高负载单原子的金属-氮-碳(M‑N‑C)的批量生产[7]。利用高温热处理碳载体、配位剂和金属盐的混合物制备得到的单金属催化剂具有优异的ORR催化性能,且具有比商业Pt/C更高的起始电位和半波电位。单金属本身具备优良的电子导电性,这在ORR催化过程中有利于电子转移。因此,通过单金属和高导电碳共同掺杂以增强硫化钴导电性具有现实意义,三者所形成的金属-硫化钴/高导电碳复合结构实现了多种活性位点的共存,这将进一步增强复合材料的ORR催化性能。
我们利用富含过渡金属钴、氮、碳元素的普鲁士蓝类似物(Co‑Co PBA)作为单金属钴的前驱体,与钴盐和氮掺杂科琴黑(N‑KB)在混合溶剂中共同硫化,后期直接高温煅烧以制备原位N‑KB负载的金属钴-硫化钴纳米颗粒(Co‑Co9S8/N‑KB)。在ORR过程中,其以4电子机制催化ORR,总体催化性能可与商业Pt/C相媲美。在自制铝-空气电池的应用中,其全电池性能表现突出,具有较高的放电电压。
1 实验部分
1.1 Co⁃Co PBA前驱体的合成
在已有文献[8‑9]报道的基础上,经过部分改动,利用液相共沉淀法成功制备了Co‑Co PBA。具体方法如下:在室温下,将2 mmol钴氰化钾溶于100 mL去离子水中,标记为A液;然后将3 mmol硝酸钴和4.5 mmol柠檬酸钠溶于100 mL去离子水中,磁力搅拌10 min,标记为B液。利用注射器将A液缓慢匀速地滴入B液中,滴入时间约30 min,在滴入过程中保持磁力搅拌。随后,将所得混合液静置老化24 h。待老化结束后,离心洗涤数次,离心速度10 000 r·min-1,时间10 min。最后,将收集到的产物置于真空干燥箱中60℃干燥过夜,所得粉红色粉末即为Co‑Co PBA纳米粒子。
1.2 Co⁃Co9S8/N⁃KB催化剂的合成
N‑KB的合成:准确称取0.2 g KB(KB600)和1.2 g三聚氰胺分散于80 mL去离子水中,超声处理30 min。将其快速转移到100 mL反应釜中,在120℃下连续反应24 h。待反应釜自然冷却至室温后,利用0.15 μm孔径的滤膜进行抽滤,之后,置于80℃下真空干燥过夜。将干燥所得物充分研磨后,转置于瓷舟,在瓷舟外用铜箔包裹及铜丝捆绑。将处理好的瓷舟放入管式炉中,在氩气气氛下以5℃·min-1的速率升至650℃后保持2 h。自然冷却至室温,所得样品即为N‑KB。
Co‑Co9S8/N‑KB催化剂的合成:准确称取200 mg N‑KB,超声分散于20 mL去离子水中。加入500 mg Co‑Co PBA,继续超声30 min。加入1.7 mmol硫酸钴和1.5 mmol硫脲,搅拌使其溶解。加入20 mL乙二醇(EG)和20 mL N,N‑二甲基甲酰胺(DMF)混合液,搅拌10 min。将其转移到100 mL反应釜中,在鼓风干燥箱中180℃持续反应12 h。反应结束后,自然冷却至室温,多次离心洗涤(10 000 r·min-1,10 min)并收集产物,将所收集物置于60℃下真空干燥。最后,在氩气气氛下,以5℃·min-1的速率升至800℃并煅烧2 h,所得产物命名为Co‑Co9S8/N‑KB。
为了对比,我们采用上述相同方法合成了Co/N‑KB和Co9S8/N‑KB。对于Co/N‑KB而言,在合成部分没有加入硫脲;对于Co9S8/N‑KB而言,在合成部分没有加入Co‑Co PBA。此外,为了探究煅烧温度对材料ORR催化性能的影响,我们还在600、700和900℃分别进行煅烧处理得到相应的Co‑Co9S8/N‑KB。
1.3 物理表征方法
采用X射线衍射仪(XRD,UltimaⅣ,Rigaku,Japan)对样品的物相进行表征,测试条件:Cu靶Kα射线(λ=0.154 nm),加速电压为 40 kV,电流为 40 mA,扫描范围 10°~90°,扫速为 5(°)·min-1。采用傅里叶变换红外光谱仪(FT‑IR,Nicolet 380,Thermo‑Electron,USA)对样品的分子结构和化学组成进行分析。采用热重分析仪(TG,Q50,TA,USA)对材料的热性能进行分析。采用X射线光电子能谱仪(XPS,Escalab 250 XI,ThermoFisher,USA)对样品表面化学元素及价态进行分析。采用扫描电镜(SEM,Nova Nanosem 230,FEI,USA)和透射电镜(TEM,Titan G2 60‑300,FEI,USA)对材料的微观形貌和微观结构进行分析,其工作电压(分辨率)分别为30 kV(1 nm)及300 kV(80 pm)。
1.4 电化学性能测试
1.4.1 电极的制备
准确称取2 mg样品和4 mg N‑KB于4 mL的样品管中,加入950 μL无水乙醇,超声20 min得到均匀的分散液,再加入50 μL Nafion(质量分数5%)黏结剂,继续超声20 min得到均匀的电极墨汁。用移液枪准确量取8 μL电极墨汁滴涂在RDE或RRDE(旋转盘电极或旋转环盘电极)工作电极上,在红外灯下干燥5 min后待测。为了对比,称取6 mg商业级Pt/C(质量分数20%),不加N‑KB,按照上述步骤制备电极。所制电极的活性物质负载量(以总质量6 mg来计算)均为0.244 5 mg·cm-2。
铝-空气电池的组成:负极为金属铝条(2 cm×10 cm),正极为催化层,电解质为含有6 mol·L-1KOH、0.01 mol·L-1Na2SnO3、0.000 5 mol·L-1In(OH)3和0.007 5 mol·L-1ZnO的混合溶液。其中,催化层的制备如下:称取60 mg(Pt/C为60 mg;其他为20 mg样品+40 mg N‑KB)催化剂于玛瑙研钵中,加入少量无水乙醇,持续研磨直至变成均匀浆体,加入50 mg质量分数60%的聚四氟乙烯乳液(PTFE)作为黏结剂,用玻璃棒将其擀成2 cm×2 cm正方形片。最后,采用热压法将此正方形片和防水透气碳布分别压在泡沫镍两侧,形成薄片正极,并在80℃真空干燥12 h,待用。
1.4.2 性能测试
采用上海辰华CHI760E电化学工作站和美国Pine公司生产的旋转环盘电极装置对所制电极进行ORR性能测试。测试在三电极体系下进行,其中对电极为铂丝电极,参比电极为Hg/HgO电极,工作电极为表面涂覆催化剂的RDE或RRDE。
在进行电化学测试之前,先在电解液中通30 min的高纯氧气使其达到氧饱和状态。在400、625、900、1 225、1 600 r·min-1的转速及10 mV·s-1的扫描速率下分别进行线性扫描伏安(LSV)测试;RRDE的环电压设置为0.1 V。循环伏安(CV)测试时的扫描速率与LSV一致,转速为0。计时电流(i‑t)测试的电压为 0.5 V,转速为 400 r·min-1,持续时长为 10 000 s。加速老化实验是在0.5~1.0 V电压范围内进行,在氧饱和的0.1 mol·L-1KOH溶液中进行5 000次CV循环,比较循环前后LSV曲线半波电位的变化。
为了方便对比,本研究中的电位值均被转换成可逆氢电极电位(ERHE):

其中EHg/HgO为参比电极电位;0.098为25℃下参比电极的标准电位;pH为电解液(0.1 mol·L-1KOH)的酸碱度值。
利用Koutecky‑Levich(K‑L)曲线来表示j-1和ω-1/2之间的线性关系。通过K‑L曲线斜率可以计算出电子转移数(n),相应计算公式如下:
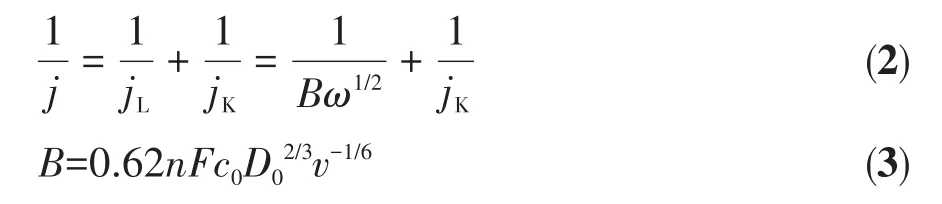
其中,j是实验中测得的电流密度,jL是扩散极限电流密度,jK是动力学极限电流密度,ω是电极转速(以角速度计),B为K‑L曲线的斜率,F为法拉第常数(96 485 C·mol-1),c0为 0.1 mol·L-1KOH 中 O2的总体浓度(1.2×10-6mol·cm-3),D0为在 0.1 mol·L-1KOH 中O2的扩散系数(1.9×10-5cm2·s-1),v为电解质的运动黏度(0.01 cm2·s-1)。
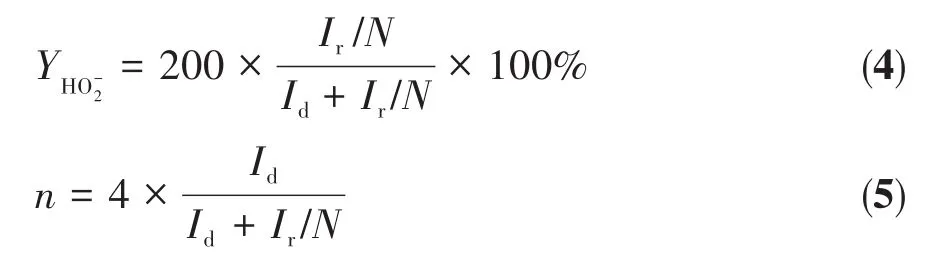
其中,Id为盘电流,Ir为环电流,N为Pt环(RRDE上的铂环)的收集效率(N=0.37)。
2 结果与讨论
2.1 Co⁃Co PBA前驱体的表征与分析
Co‑Co9S8/N‑KB纳米颗粒的制备过程详见图1。简而言之,利用 K3[Co(CN)6]、Co(NO3)2、C6H5Na3O7得到规整的Co‑Co PBA立方体颗粒。再将Co‑Co PBA与N‑KB、硫酸钴、硫脲混合,经水热反应、惰性气氛保护煅烧,最终形成球形纳米颗粒状Co‑Co9S8/N‑KB产物。
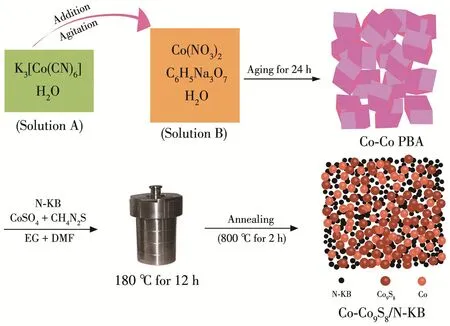
图1 Co‑Co9S8/N‑KB纳米颗粒的制备过程示意图Fig.1 Schematic illustration of Co‑Co9S8/N‑KB nanoparticles
图2a为Co‑Co PBA前驱体的微观形貌图,可以发现,该前驱体呈现出规整的三维立方体结构,尺寸均一,约0.8 μm×0.8 μm×1.7 μm。图2b为Co‑Co PBA的XRD图,结果表明,所有衍射峰都与标准卡片(PDF No.71‑0807)完美匹配,对应于[Co3(Co(CN)6)2(H2O)12]1.333物相[10]。此外,衍射峰清晰、尖锐,无杂峰,表明[Co3(Co(CN)6)2(H2O)12]1.333颗粒尺寸较小且纯度较高。图2c是Co‑Co PBA前驱体的FT‑IR谱图。在2 173 cm-1处的尖峰可归因于CN-伸缩振动,在3 645 cm-1处的ν(O—H)和1 611 cm-1处的δ(O—H)表明了水分子的存在,而在指纹区456 cm-1处的峰揭示了不同价态钴的细微变化[10]。对于3 420 cm-1处的吸收峰,可归因于水分子缔合或氰基与水分子间形成的N—H氢键的伸缩振动。
对Co‑Co PBA前驱体进行了TG分析,测试条件为在氮气气氛下以10℃·min-1升温速率从室温升至700℃,结果如图2d所示。由该图可知,Co‑Co PBA的热失重过程包含了2个阶段:在200℃之前,结晶水大幅度挥发(约21.3%);在350~600℃内,有机配体、氰化物裂解(约27.8%)。当温度超过600℃时,质量损失较小,但仍然存在。因此,我们探讨了不同煅烧温度(600、700、800和900℃)对产物催化性能的影响。
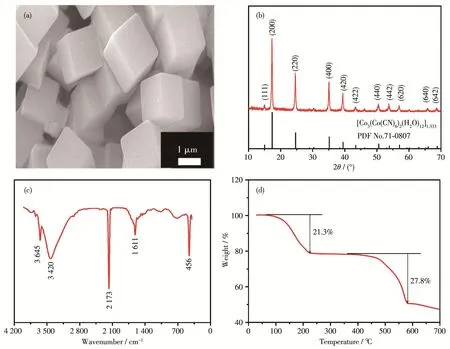
图2 Co‑Co PBA前驱体的(a)SEM图、(b)XRD图、(c)FT‑IR图和(d)TG曲线Fig.2 (a)SEM image,(b)XRD pattern,(c)FT‑IR spectrum and(d)TG curve of Co‑Co PBA precursor
2.2 Co⁃Co9S8/N⁃KB催化剂的表征
图3为N‑KB、Co9S8/N‑KB、Co‑Co9S8/N‑KB和Co/N‑KB的XRD图。如图所示,N‑KB在2θ=24°显示一个宽峰,这是典型石墨化碳(200)晶面衍射峰[11‑12]。值得注意的是,该宽峰在Co9S8/N‑KB、Co‑Co9S8/N‑KB和Co/N‑KB的XRD图中几乎不存在,这主要是由于金属Co和Co9S8的强衍射峰将其弱化。对于Co9S8/N‑KB而言,在 29.3°、31.2°、47.5°和52.1°出现的衍射峰可以很好地索引到Co9S8物相(PDF No.65‑6018)的(311)、(222)、(511)和(440)晶面。在 Co‑Co9S8/N‑KB中,可明显观察到Co/N‑KB和Co9S8/N‑KB各自的衍射峰,这表明了单金属Co被成功地引入到N‑KB支撑的Co9S8相中,单金属Co的存在不仅可作为催化活性位点,而且还能增强Co‑Co9S8/N‑KB复合材料的导电性,加快ORR催化过程中电子转移速率,促进其电化学活性。而在44.2°、51.5°和75.8°处出现的尖锐衍射峰与金属Co标准物相卡片(PDF No.15‑0806)一致,且分别对应(111)、(200)和(220)晶面。值得注意的是,根据Co9S8/N‑KB和Co/N‑KB的合成原料差异可知,在未加入Co‑Co PBA时产物为Co9S8/N‑KB,而在未加入硫脲时产物为Co/N‑KB。这可证明,在Co‑Co9S8/N‑KB中各项组分来源不同,金属Co主要来源于Co‑Co PBA的高温裂解,而Co9S8是由硫酸钴和硫脲结合所得。

图3 N‑KB、Co9S8/N‑KB、Co‑Co9S8/N‑KB和Co/N‑KB的XRD图Fig.3 XRD patterns of N‑KB,Co9S8/N‑KB,Co‑Co9S8/N‑KB and Co/N‑KB
图4为Co‑Co9S8/N‑KB表面元素组成和化学价态的表征结果。由图4a可知,Co‑Co9S8/N‑KB主要由C、Co、S、N和O五种元素组成,其分别为86.91%、1.23%、0.99%、4.3%和6.57%。从图4b可知,C元素可拟合成4个峰,分别位于284.75、285.60、287.85和290.10 eV,对应于C=C/C—C、C—N/C—S、C=O和O—C=O[13]。图4c为Co2p高分辨XPS谱图,其中位于780.41、795.30和796.79 eV处的尖锐峰分别对应于Co2p3/2(难以再分峰)、Co2+(Co2p1/2)和Co3+(Co2p1/2)[13],而在785.52和802.53 eV处的2个峰对应2个卫星峰[13‑14]。图4d为S2p的高分辨XPS谱图,位于结合能163.85 eV处的峰对应于S2-,是典型的Co—S键,而位于165.20和168.52 eV处的峰对应于S=C和SO42-键[15]。N1s高分辨XPS谱图可以很好地拟合成3个峰 (图 4e),分 别 为 吡 啶 氮 (398.54 eV)、石 墨 化 氮(401.15 eV)和氧化氮(405.41 eV)[15]。可以发现,石墨化氮的含量远高于其他2种氮,石墨化氮稳定且导电性高,这有利于增强材料导电性,促进ORR催化性能[15]。图4f是O1s的高分辨XPS谱图,从图中可以得到在结合能530.15 eV处有一个Co—O键,这主要由于材料表面的Co单金属与空气接触而被氧化。而位于结合能531.30和532.60 eV处的峰对应于C=O和物理吸附水,这有利于催化剂材料在电解液中的浸润[16‑17]。
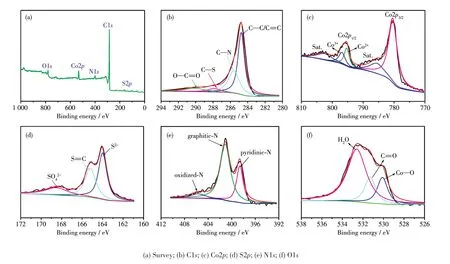
图4 Co‑Co9S8/N‑KB的XPS谱图Fig.4 XPS spectra of Co‑Co9S8/N‑KB
采用SEM、TEM技术对Co‑Co9S8/N‑KB的微观形貌和微观结构进行了全面表征(图5)。由图5a~5c可知,Co‑Co9S8/N‑KB为直径约50 nm的球形纳米颗粒。在图5d的TEM图中可观察到一些颜色深暗的颗粒(23~50 nm),这些颗粒被证实为金属Co和Co9S8颗粒(图5e、5f)。在高分辨TEM图(图5e、5f)中,晶格间距为0.291、0.193和0.182 nm的晶格条纹分别对应于Co9S8的 (311)、(511)和 (440)晶 面 ,而 晶 格 间 距 为0.209、0.136和0.178 nm处的晶格条纹对应于金属Co的(111)、(220)和(200)晶面。这有力地证实了金属Co和Co9S8共存于该材料中,这必将大大提高其电化学性能。

图5 Co‑Co9S8/N‑KB的SEM图 (a~c)、TEM图 (d)和HRTEM图 (e、f)Fig.5 SEM images(a~c),TEM image(d)and HRTEM images(e,f)of Co‑Co9S8/N‑KB
为了进一步研究Co‑Co9S8/N‑KB的元素组成和分布情况,我们进行了扫描透射电子显微镜-能量色散X射线谱(STEM‑EDS)分析(图6)。从图6可以看出Co、S、N、C、O元素共同存在于Co‑Co9S8/N‑KB中,这与XPS(图4)结果高度一致。此外,还可以清楚地观察到Co、S、N元素比较均匀地分布在N‑KB中,形成了氮掺杂碳支撑的高导电性结构,为电子转移提供了便利,这将有利于其催化性能的提高。
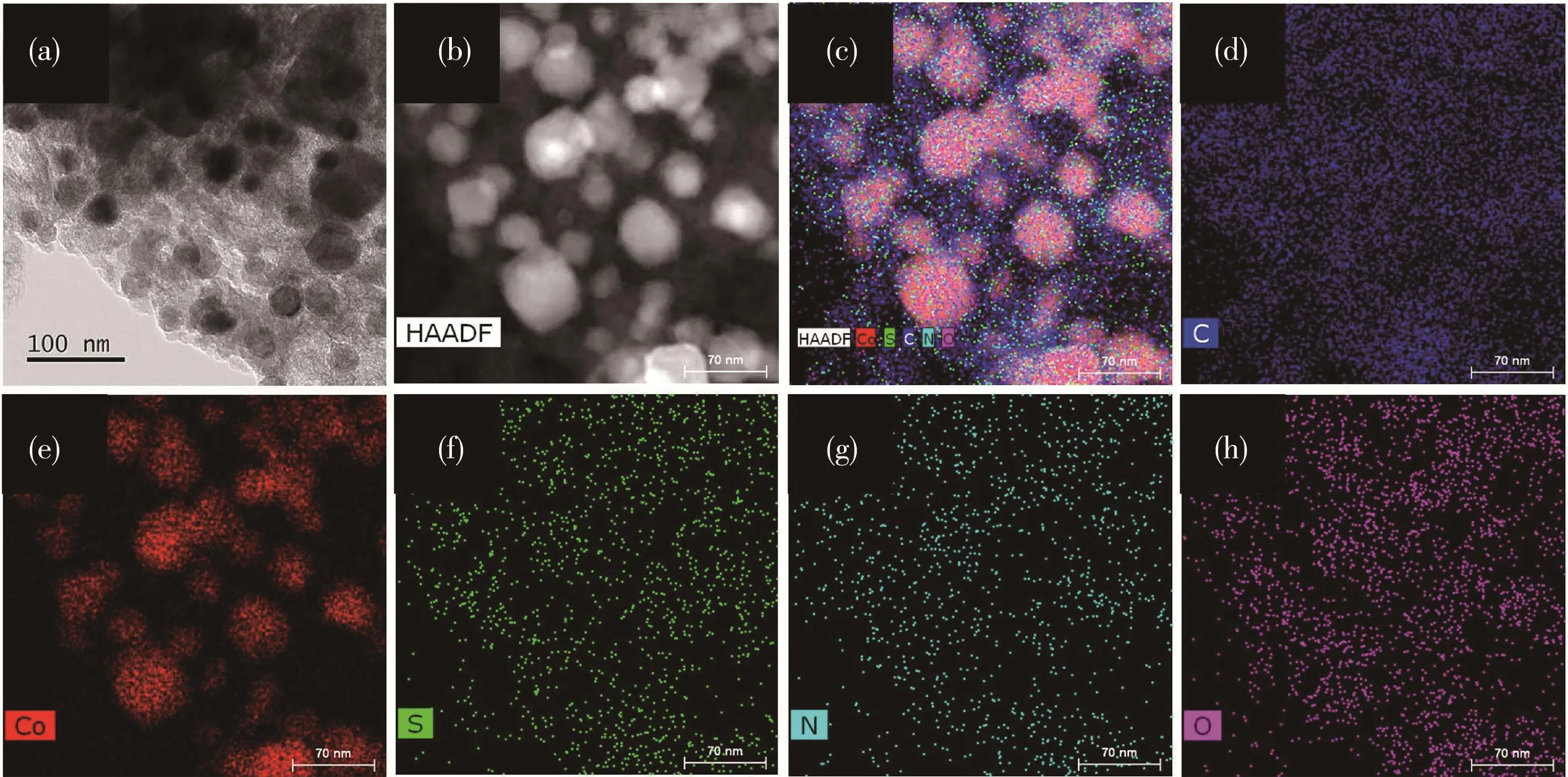
图6 Co‑Co9S8/N‑KB的(a)TEM图、(b)HADDF图和(c~h)STEM‑EDS图Fig.6 (a)TEM image,(b)HADDF image and(c~h)STEM‑EDS mappings of Co‑Co9S8/N‑KB
2.3 Co⁃Co9S8/N⁃KB催化剂的电化学性能分析
图7a为不同样品在氧饱和的0.1 mol·L-1KOH溶液中,1 600 r·min-1转速下的LSV极化曲线。如图所示,未进行氮掺杂的KB催化剂表现出非常低的ORR活性,而利用三聚氰胺作为氮源进行掺杂的N‑KB催化剂活性有所提升。利用Co‑Co PBA前驱体经热解制得的Co/N‑KB以及利用钴离子直接硫化形成的Co9S8/N‑KB,都显示出比N‑KB更好的催化性能,这表明金属钴粒子、Co9S8活性颗粒与高导电N‑KB之间都具有协同催化效应。值得注意的是,将单金属Co引入到Co9S8/N‑KB中所形成的Co‑Co9S8/N‑KB具有非常优异的催化性能,表现出与商业级Pt/C相近的起始电位和半波电位,分别约为0.93和0.83 V。此外,Co‑Co9S8/N‑KB的极限电流密度为5.7 mA·cm-2,超过了商业级Pt/C(5.3 mA·cm-2)。这可能是由于在Co‑Co9S8/N‑KB中,单金属Co的存在能进一步增强材料的导电性,使得在催化过程中电子转移速率更快,而Co9S8颗粒提供足够的活性位点,能更有效地催化ORR[18]。图7b为在氧饱和氛围下不同催化剂的CV测试图。从该图可以观察到,所有样品在0.8 V左右都有一个强还原峰,这表明这些样品均能对ORR起较强的催化作用。
为了探究催化剂在催化ORR过程中的动力学特征,在不同转速下对Co‑Co9S8/N‑KB、商业级Pt/C进行了LSV测试,并绘制了K‑L曲线,如图7c、7d所示。结果发现,在不同电压下,Co‑Co9S8/N‑KB、Pt/C的K‑L曲线中j-1与ω-1/2均具有良好的线性关系,这表明电解质中所溶解的氧呈现出一级反应动力学特征[19‑20]。根据K‑L方程计算了Co‑Co9S8/N‑KB、Pt/C催化ORR的电子转移数,发现在0.1~0.6 V范围内Co‑Co9S8/N‑KB催化ORR的电子转移数为3.89~3.94(图7c),Pt/C的为3.94~3.98(图7d),都接近于4电子,表明Co‑Co9S8/N‑KB具有与Pt/C相似的动力学过程,预示了其高效的ORR催化性能[21]。图7e是不同催化剂的Tafel曲线,可以看出Co‑Co9S8/N‑KB的Tafel曲 线 斜 率 为 81.9 mV·dec-1,优 于 Pt/C(82.4 mV·dec-1)、Co/N‑KB(88.6 mV·dec-1)、Co9S8/N‑KB(86.3 mV·dec-1),证实了其具有最佳的ORR活性[22]。由图7f可知,在0~0.8 V电位下Co‑Co9S8/N‑KB与Pt/C催化剂的过氧化物产率都低于10%,电子转移数都接近4,与图7c结果高度一致,进一步证明了Co‑Co9S8/N‑KB具有高效的4电子转移机制。

图7 样品的电化学性能:(a)LSV曲线;(b)CV曲线;(c)Co‑Co9S8/N‑KB的LSV曲线和K‑L曲线;(d)Pt/C的LSV曲线和K‑L曲线;(e)Tafel曲线;(f)和nFig.7 Electrochemical properties of samples:(a)LSV curves;(b)CV curves;(c)LSV curves of Co‑Co9S8/N‑KB and K‑L plots;(d)LSV curves and K‑L plots of Pt/C;(e)Tafel plots;(f)and n
图8为不同煅烧温度下所得Co‑Co9S8/N‑KB复合材料在1 600 r·min-1转速下的LSV曲线。由图可知,在800℃下进行煅烧所得的Co‑Co9S8/N‑KB表现出最优性能,与Pt/C相当。当温度较低(600、700℃)时性能不佳,可能是因为前驱体未能充分热解还原成单金属Co或者氮原子石墨化程度不够;温度过高(900℃)则可能会使材料中氮含量降低,从而降低了其催化活性。
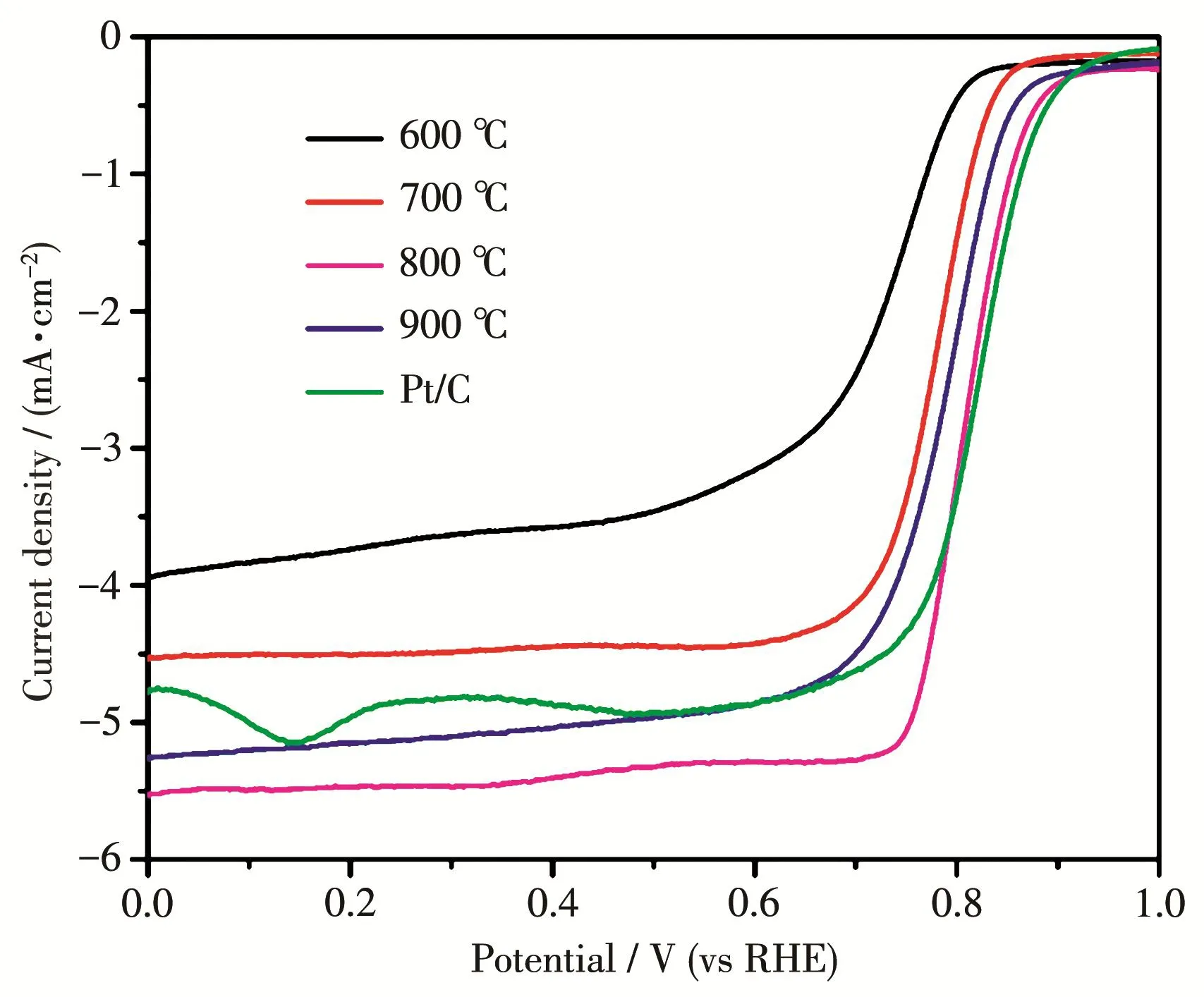
图8 不同煅烧温度下制得的Co‑Co9S8/N‑KB催化剂的LSV曲线Fig.8 LSV curves of Co‑Co9S8/N‑KB catalysts prepared at different annealing temperatures
催化剂的稳定性也是判断其是否具备实际应用价值的一个重要指标,为此,我们采用计时电流法及加速老化实验对Co‑Co9S8/N‑KB的稳定性进行了测试(图9)。在电位保持为0.5 V、转速为400 r·min-1的条件下对Co‑Co9S8/N‑KB进行计时电流测试图(9a),结果发现,在经过10 000 s长时间测试之后,电流密度还能保持在初始值的95.6%,优于Pt/C(88.7%,图9c),这证明其具备优良的稳定性。为了进一步验证该催化剂稳定性,我们对其在0.5~1.0 V电压范围内进行了加速老化实验,比较了5 000次CV循环前后半波电位的变化(图9b)。从图9b可知,在5 000次CV循环后,Co‑Co9S8/N‑KB的半波电位(ΔE1/2)仅负移了8.6 mV,优于Pt/C(34.1 mV,图9d),与文献结果相似[23],这进一步证实了Co‑Co9S8/N‑KB催化剂具有卓越的催化活性稳定性。
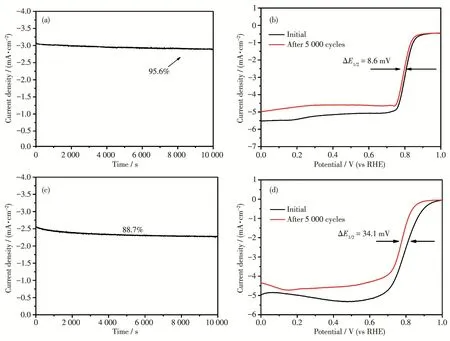
图9 Co‑Co9S8/N‑KB的计时电流曲线 (a);Co‑Co9S8/N‑KB循环5 000次前后的LSV曲线 (b);Pt/C的计时电流曲线(c);Pt/C循环5 000次前后的LSV曲线(d)Fig.9 Chronoamperometric curves of Co‑Co9S8/N‑KB(a);LSV curves of Co‑Co9S8/N‑KB before and after 5 000 cycles(b);Chronoamperometric curve of Pt/C(c);LSV curves of Pt/C before and after 5 000 cycles(d)
图10为铝-空气电池的放电性能测试图。图10a为在50 mA·cm-2恒定电流密度下所得的放电曲线,由该图可知,待阳极铝完全活化后,Co‑Co9S8/N‑KB最终可达1.53 V的工作电压,高于Pt/C(1.43 V)。图10b为不同放电电流密度下对应的输出电压曲线,由该图可知,两者的输出电压都随放电电流密度增加而降低,但不同放电电流密度下Co‑Co9S8/N‑KB的输出电压明显高于Pt/C,具有更好的全电池实际应用效果。
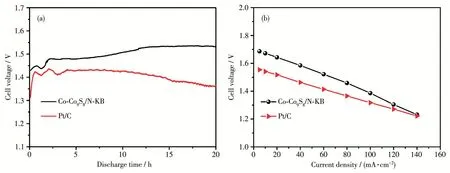
图10 (a)全电池恒流放电曲线;(b)全电池极化曲线Fig.10 (a)Constant current discharge curves of the full batteries;(b)Polarization curves of the full batteries
3 结 论
以自制普鲁士蓝类似物Co‑Co PBA为前驱体,通过水热反应和高温煅烧过程合成了高导电碳支撑、单金属Co引入的Co‑Co9S8/N‑KB复合ORR催化剂。单金属Co的引入增强了材料导电性,能加快ORR催化过程中电子的转移速率,从而使Co‑Co9S8/N‑KB表现出十分优异的电化学性能,具备与商业Pt/C相近的起始电位和半波电位以及更大的极限电流密度。Co‑Co9S8/N‑KB在催化稳定性方面也表现突出,在经过10 000 s长时间持续催化后,其电流密度还能保持95.6%;在动态循环5 000次后,其半波电位仅偏移了8.6 mV。将该催化剂用作铝-空气电池的阴极催化剂,在50 mA·cm-2电流密度下进行全电池放电实验时,表现出比商业Pt/C更高的放电电压平台,高达1.53 V。本项工作为合成单金属引入的复合型ORR催化剂提供了一种简便有效的策略,所合成Co‑Co9S8/N‑KB催化剂性能优良,有望替代Pt/C作为铝-空气电池阴极催化材料。
猜你喜欢
蓄电池(2022年1期)2022-02-25
陶瓷学报(2020年2期)2020-10-27
陶瓷学报(2020年2期)2020-10-27
中学生数理化(高中版.高二数学)(2020年2期)2020-04-21
商品与质量(2019年42期)2020-01-17
天津医科大学学报(2019年3期)2019-08-13
中国资源综合利用(2017年4期)2018-01-22
中国有色冶金(2016年3期)2016-02-11
中国资源综合利用(2016年4期)2016-01-22
科技资讯(2015年8期)2015-07-02
