
基于Rietveld精修方法解析Bi2O3的原子热振动
2021-09-22 07:32王丹丹刘泽朋
人工晶体学报 2021年8期
王丹丹,刘泽朋,香 莲
(内蒙古民族大学数理学院,通辽 028043)
0 引 言
Bi2O3是氧离子(O2-)导电体,它是一种先进的功能材料,也是最重要的铋化合物之一。近年来,因其具有大的能带隙、折射率、介电常数以及显著的光电导和光致发光等光学和电学性质而被广泛应用于电子陶瓷材料、电解质材料、光电材料、传感器、微电子元件、高温超导材料、催化剂、铁电材料等各领域中,同时还用于化学试剂、铋盐、防火材料、高折光率玻璃、核工程玻璃制造和核反应堆燃料等方面[1]。Polat等[2]制备了Gd2O3和Lu2O3双掺杂Bi2O3化合物,表明了稳定的δ-Bi2O3相样品可用作固体氧化物燃料电池的电解质材料。丁鹏等[3]制备了Bi2O3-Ni2O3纳米粉末,经750 ℃焙烧的光催化剂对苯光催化降解活性最高。Hakimi等[4]通过非水溶胶-凝胶路线合成Bi2O3-Al2O3纳米复合材料,显示出比Al2O3更高的光催化活性。Viruthagiri等[5]用一种新的化学沉淀方法来制备不同浓度的钴掺杂Bi2O3纳米颗粒,表现出优异的结构和光学性质。Raju等[6]研究了ZnO-Bi2O3-Yb2O3基压敏陶瓷,提高了压敏电阻组件在侵入纳秒瞬态下从非导电态转变到导电态的转变速度。徐东等[7]制备了掺Lu2O3的ZnO-Bi2O3基压敏陶瓷样品,具有比较理想的综合电性能。熊智慧等[8]采用第一性原理杂化泛函HSE06方法对Fe掺杂α-Bi2O3的电子结构和光学性质进行了计算研究,为Fe掺杂α-Bi2O3在光催化领域中的应用提供了理论依据。Huang等[9]通过溶剂热制备和后退火处理来修饰Eu3+掺杂的铋氧化物的相形成、形态特征和光能转化,提出了光能转换机理来讨论发光性质、衰变寿命和多价离子。三氧化二铋(Bi2O3)有七种晶相,分别为单斜α相(α-Bi2O3)、四方β相(β-Bi2O3)、体心立方γ相(γ-Bi2O3)、面心立方δ相(δ-Bi2O3)、正交ε相(ε-Bi2O3)、三斜ω相(ω-Bi2O3)和六方η相(η-Bi2O3)。其中最稳定的结构为α相和δ相,亚稳定结构为β、γ、ε和η相,而ω相因极不稳定而存在争议[10]。本研究通过X射线对粉末晶体Bi2O3进行衍射实验,利用Rietveld解析方法中的RIETAN-2000程序[11]对实验结果进行全谱拟合晶体结构精修,得到原子热振动各向同性因子B。B是与材料热性质相关的重要参数,利用它可计算出德拜-劳厄因子,也可推导出原子间热振动相关效果μ的值,并进一步计算出比热和色散谱[12],原子热振动大小的B值与温度有关,且随温度增高而增大[13-14]。用最大熵方法(maximum entropy method, MEM)解析方法通过Practice Iterative MEM Analyses(PRIMA)模块和Vi-sualization of Electron/Nuclear Densities(VEND)模块[15-19]进行等高电子密度分布可视化,确定晶体结构和原子的位置。
1 Rietvld精修和MEM解析
1.1 Rietvld精修
Rietveld精修是由设在荷兰的Petten反应堆中心的研究员Rietveld在1967年用中子多晶体衍射数据精修晶体结构参数时提出的一种数据处理新方法——多晶体衍射全谱线形拟合法。1977年,Malmros和Thomas首次将这一方法应用到X射线领域。Rietveld全谱拟合是以一个晶体结构模型为基础,利用它的各种晶体结构参数及一个峰形函数计算一张在2θ范围内的、理论的多晶体衍射谱,将此计算谱与实验测得的衍射谱比较并修改,反复进行多次,以使计算谱和实验谱的差最小(最小二乘法),拟合的目标是整个衍射谱的线形,拟合范围是整个衍射谱,不是个别衍射峰。整个衍射谱是各衍射峰强度分布的叠加,故衍射谱上某点 (2θ)i处的实测强度Yi表示为
Yi=Yib+∑kYik
(1)
式中:Yib为本底强度。在作Yik加和时,需设定每个衍射峰的延展范围,定义为该峰的半峰宽(FWHM)的若干倍,如5倍或7倍等。这样可以知道在某个(2θ)i处的Yi是由哪几个衍射贡献的。根据一定的晶体结构模型及峰形函数,可以计算衍射谱上各(2θ)i处的衍射强度Yic(下标c表示为计算值)。改变各结构参数或峰形参数,可改变Yic。用非线性最小二乘法使Yic拟合实测的高分辨、高准确的数字多晶体衍射谱上的各Yio(下标o表示实测值),也即下式之中M最小:
M=ΣiWi(Yio-Yic)2
(2)
式中:Wi=1/Yi为基于计数统计的权重因子;Yio为实测值,在反复循环中不变;Yic是根据模型计算的,在每次参数修改后,此值均要改变。
为了判别精修中各参数的调整是否合适,设计出一些判别因子,简称R因子。
RI=Σ|Iko-Ikc|ΣIko
(3)
(4)
式中:Iko表示积分强度的实测值;Ikc表示积分强度的计算值。这两个因子是由衍射峰的积分强度计算出的,强烈依据于结构模型,是能判断结构模型是否正确的最有价值的R因子[20]。
1.2 最大熵方法
MEM起源于Jaynes引入的统计力学领域的“最大熵原理”,原本是物理学中的一个概念,用来反映某一系统的混乱程度,后被用于数学中来描述一个信息量,即信息熵,是指在推断未知分布时,只考虑已知信息,而不随意加入主观信息,信息量越大,则熵也越大[21-22]。在热力学的相关研究中表征系统的混乱程度和无序度。在信息科学中,熵是对信息的不确定度的一种测量,用来作为一个系统的信息含量的量化指标[23-24]。此方法在地球物理学科中各种数据处理中获得了跨时代的成功,已在许多科学和技术领域中采用,例如光谱分析、图像恢复、数学和物理学等,本研究将其应用到晶体结构分析上。MEM是根据有限的信息,使用信息熵推导出统计性的最准确结论的方法,1967年由J.P.伯格所提出[25]。这个方法中,要求实验中得到的信息吻合这个限制条件下得到的信息熵的最大解。最大熵原理的含义就是,在已有的条件约束下,不添加任何人为假设,使熵达到最大,即混乱程度最高[26]。对于衍射实验中,决定电子-原子核密度时,推定观察到的结构因子在误差范围内的密度分布尽量取分散值,对于没有被测定的高角度领域部分的结构因子推定为不等于零的结构因子。
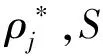
(5)
(6)
对于τj的初期值,就是熵的最大信息状态,即在体积V的单位胞中通常采用密度均衡分布的模型。在X射线衍射的情况下,如果F(000)是000反射的构造因子(等于单位胞中总电子数),那么N个像素全部设定为F(000)/V。
在衍射实验领域里MEM解析是Dilanian和泉富士夫两人开发建立的电子-原子核密度的可视化(visualization of electron/nuclear densities and structures, VENUS)程序系统。VENUS可以在三维(3D)可视化、渲染和操纵晶体结构以及各种物理量(例如电子/核密度)中发挥作用,不仅可以通过X射线和中子衍射来确定,还可以通过电子结构计算来确定,能够防止实验方法与理论方法之间的相分离[27]。
2 实 验
本研究使用的是天津市光复精细化工研究所的纯度不低于99.99%的Bi2O3粉末晶体。
首先使用型号为BT-1600的图像颗粒分析系统对Bi2O3粉末晶体进行颗粒分析,BT-1600图像颗粒分析系统包括型号为生物、金相、体视显微镜(三目),最大光学放大倍数为1 600倍的光学显微镜、500万像素的型号为RZ500C数字CCD摄像头、数据采集与处理的BT-1600软件、windows系统电脑以及彩色喷墨打印机,其重复性误差<3%(不包含样品制备因素造成的误差)。其原理为通过对颗粒数量和每个颗粒所包含的像素数量的统计,计算出每个颗粒的等圆面积和等球面积,从而得到颗粒的等圆面积直径、等球体积直径以及长径比等,再对所有的颗粒进行统计,从而得到粒度分布等信息,以微米为单位。其实验结果如表1和图1所示,实验结果表明,Bi2O3粉末晶体呈现片状,最大粒径为91.28 μm,最小粒径为0.60 μm,平均粒径为 9.42 μm,比表面积为0.092 m2/g。

图1 Bi2O3粒度分析的形状Fig.1 Shape of Bi2O3 particle size analysis

表1 室温下Bi2O3的粒度分析Table 1 Particle size analysis of Bi2O3 at room temperature
使用型号为D8 FOCUS的X射线衍射仪,对粉末晶体在室温条件下进行了衍射实验。辐射源为Cu Kα,其波长λ为0.154 nm,扫描范围10°~90°,步长0.02°,每步计数时间为3 s。实验结果如图2所示。

图2 室温下X射线衍射实验结果Fig.2 X-ray diffraction experiment results at room temperature
3 结果与讨论
室温条件下α-Bi2O3属于单斜晶系,其空间群为P21/c,每个晶胞含有8个Bi原子和12个O原子[28-29]。如图3为Bi2O3的晶体结构模型,利用RIETAN-2000程序对Bi2O3晶体结构进行了Rietveld精修,精修图谱如图4所示,实线表示计算值,点线表示 X 射线衍射实验值,短线表示峰的位置,下方的波动线是两者的差值。图中断开部分因此区间内出现了几处γ-Bi2O3的衍射峰[30]而将其删除。从精修结果可以看出,Bi2O3的实验图谱和计算值图谱均匹配得很好。

图3 Bi2O3晶体结构模型Fig.3 Bi2O3 crystal structure model

图4 Bi2O3精修图谱Fig.4 Bi2O3 refined map


表2 Bi2O3晶体结构参数Table 2 Bi2O3 crystal structure parameters
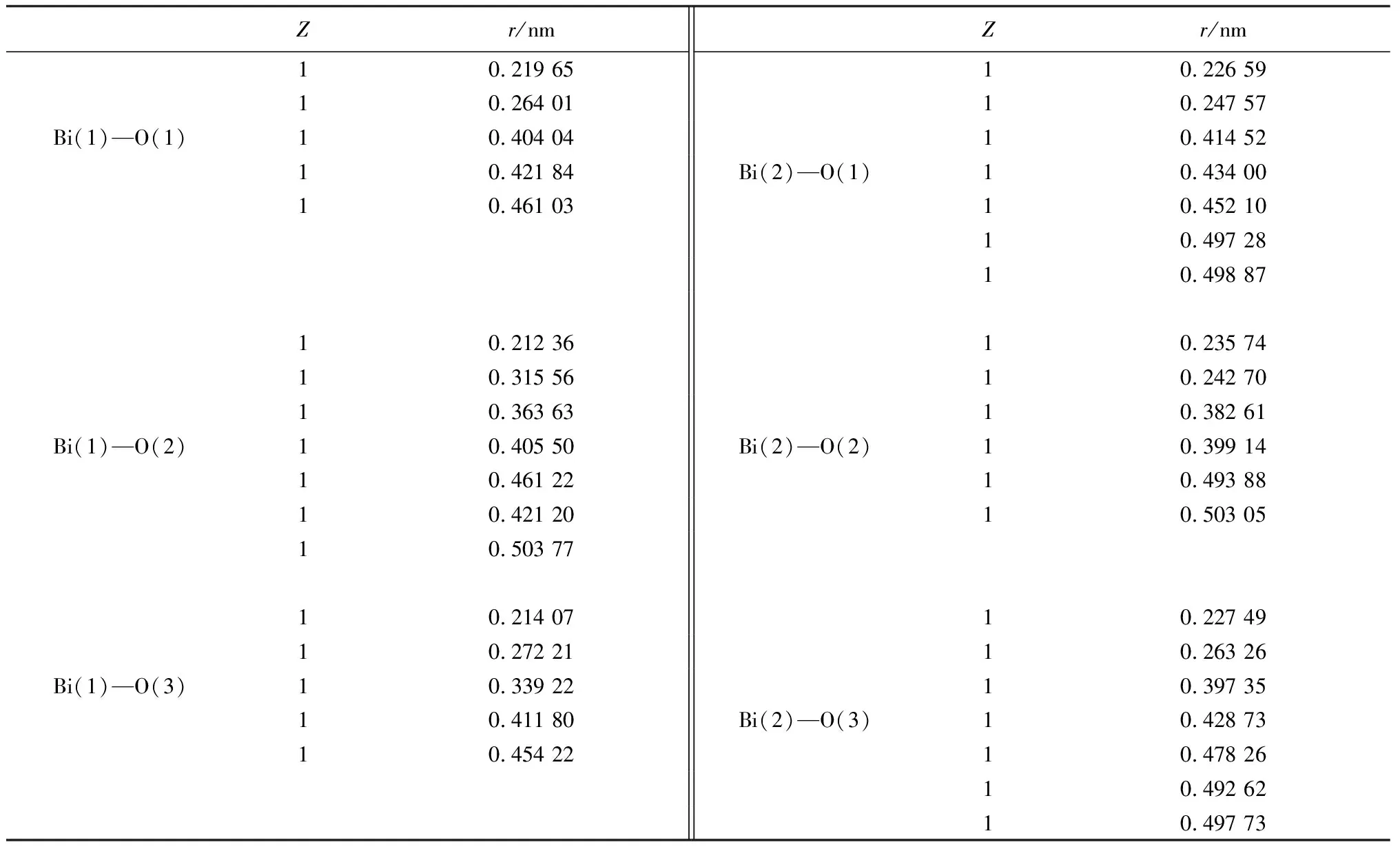
表3 Bi2O3原子配位数Z和原子距离rTable 3 Bi2O3 atomic coordination number Z and atomic distance r
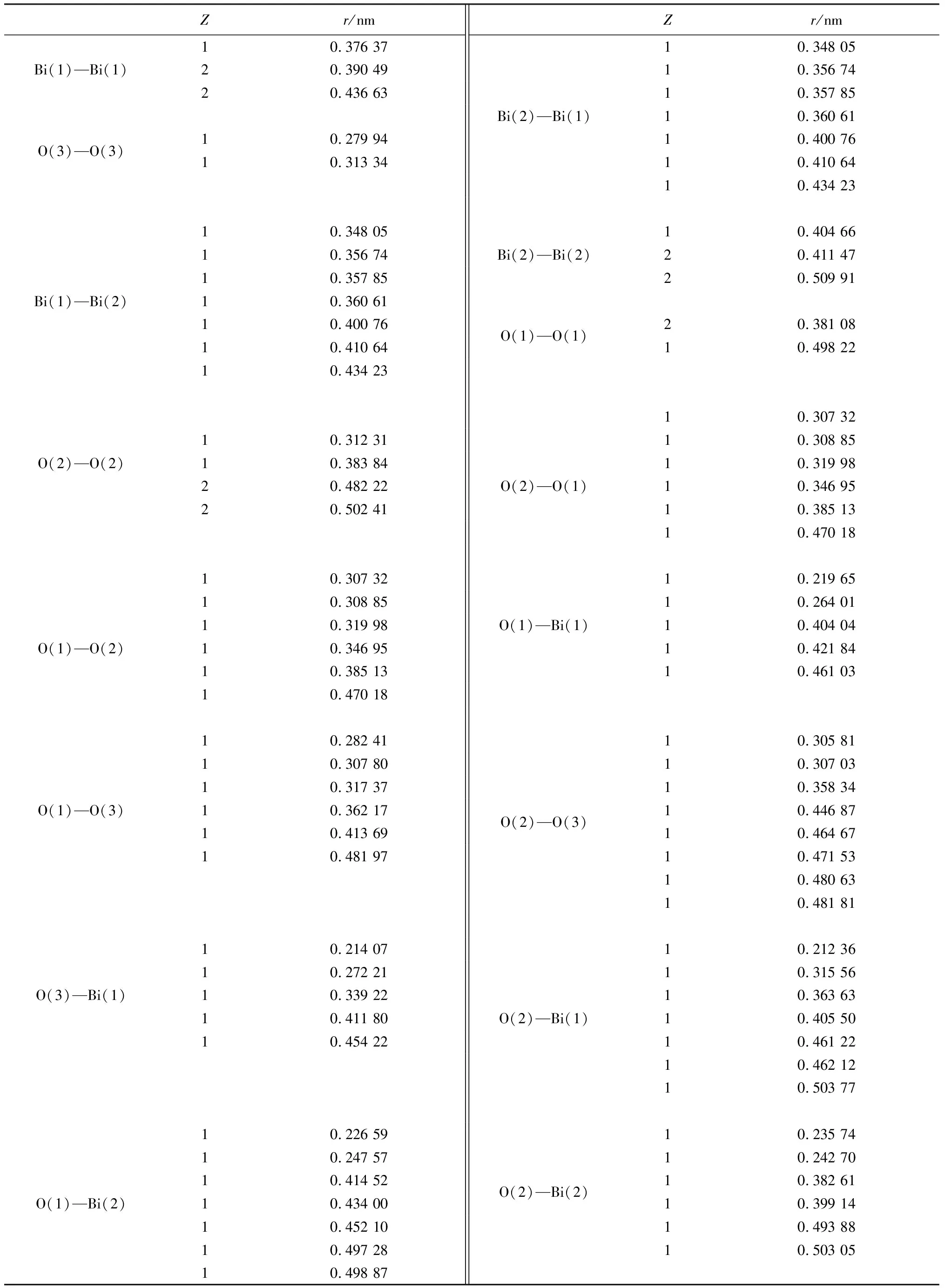
续表

续表
衍射强度公式中e-2M被称为德拜-沃勒因子(Debye-waller factor),其中M=B(sinθ/λ)2。虽然,B的数学公式已报道,但是通过公式计算极其困难,利用晶体结构精修用的Rietveld程序软件可以直接得到B的准确值。
图5和图6是采用MEM解析方法的PRIMA模块和VEND模块计算的3D(立体)和2D(平面)等高电子密度可视化分布图谱。图5、6所示的3D(立体)和2D(平面)等高电子密度分布图谱里的电子密度分布是球状分布,扩散面积小,这进一步说明了在室温附近各原子作各向同性的热振动。图5显示的3D等高电子密度分布图与图3的晶体结构模型非常符合,证明了初步建立的晶体结构模型是正确的。图6为(100)晶面的2D电子密度分布图,从立体和平面图可以看出Bi原子的确切位置,而O原子几乎看不见,这是由于Bi原子的原子序数为83,O原子的原子序数为8,X射线衍射中衍射强度与原子散射因子f有关,而原子散射因子f的大小与原子序数有关,原子序数越大原子散射因子f越大,所以重原子很容易被X射线观察到,而轻原子不容易被观察到。
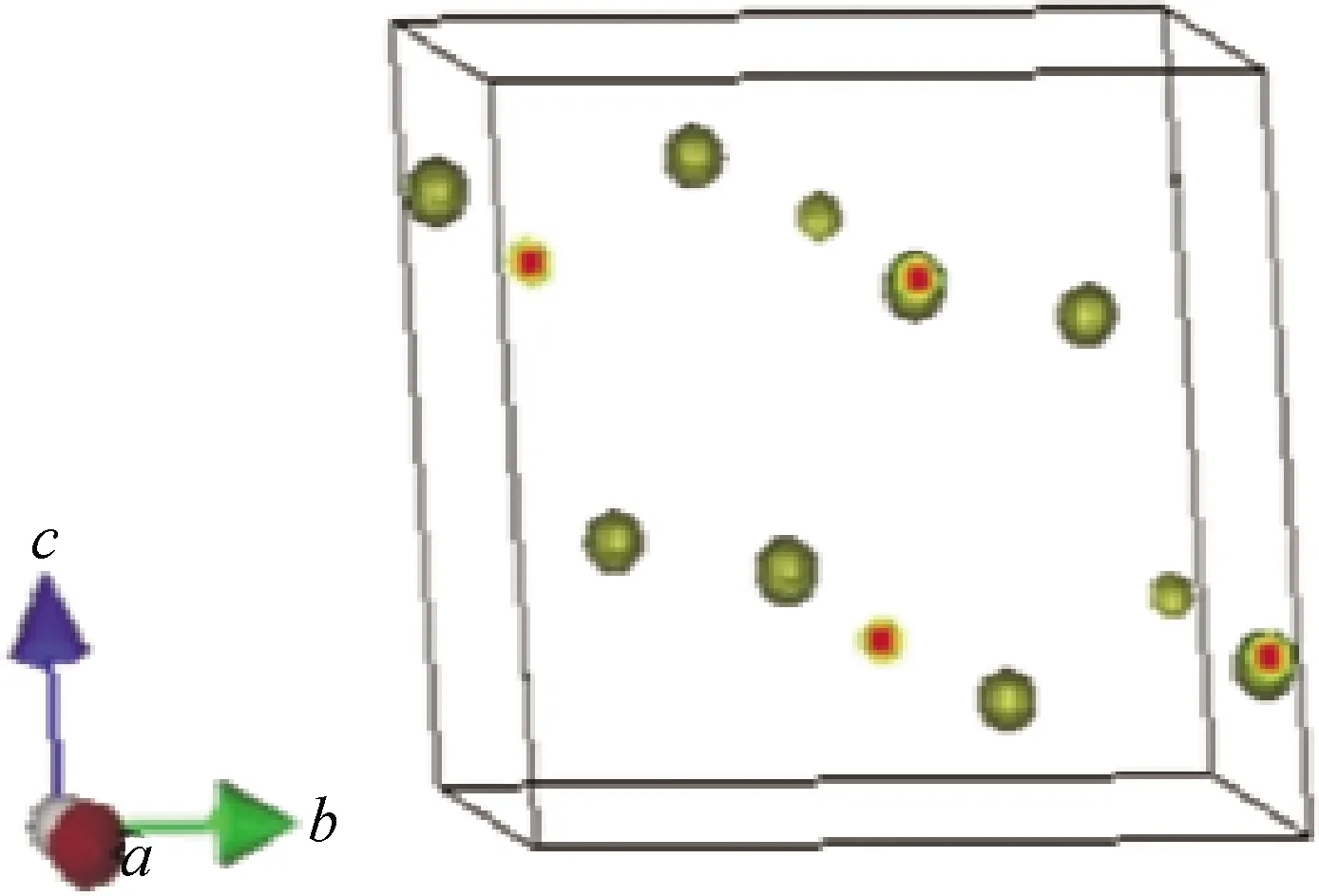
图5 3D电子密度可视化图谱Fig.5 3D electron density visualization map

图6 (100)晶面的2D电子密度可视化图谱Fig.6 2D electron density visualization map of the (100) crystal plane
4 结 论
本研究首先对Bi2O3粉末晶体进行颗粒分析,发现Bi2O3粉末晶体呈现片状,最大粒径为91.28 μm,最小粒径为0.60 μm,平均粒径为 9.42 μm,比表面积为0.092 m2/g。然后对Bi2O3粉末的晶体结构进行了精修,得到了原子热振动各向同性因子B的大小,各原子Bi(1)、Bi(2)、O(1)、O(2)和O(3)的原子热振动各向同性温度因子分别为0.004 938 nm2、0.004 174 nm2、0.007 344 nm2、0.007 462 nm2和0.007 857 nm2。B是与材料热性质相关的重要参数,利用它可计算出德拜-劳厄因子,也可推导出原子间热振动相关效果μ的值,并进一步计算出比热和色散谱。所以进一步的研究目标是结合定性和定量分析探究离子导电率和原子热振动之间的关系。通常,原子热振动各向同性温度因子B的计算比较复杂,可以通过X射线衍射实验与晶格精修方法结合直接得出。
猜你喜欢
机电安全(2022年5期)2022-12-13
中学生数理化(高中版.高考数学)(2020年2期)2020-04-21
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21
科学(2020年1期)2020-01-06
测绘通报(2019年11期)2019-12-03
赤峰学院学报·自然科学版(2019年5期)2019-09-10
科教导刊·电子版(2018年13期)2018-07-31
科技与创新(2015年21期)2015-12-01
中国卫生(2015年12期)2015-11-10
中国卫生(2015年10期)2015-11-10
