
钒改性对铁基脱硝催化剂活性及抗碱性能的影响
2021-09-22 07:32:20蔡思翔
人工晶体学报 2021年8期
李 悦,姜 宏,蔡思翔
(1.海南大学材料与科学工程学院,海口 570228; 2.海南大学,海南省特种玻璃实验室,海口 570228; 3.海南大学,南海海洋资源利用国家重点实验室,海口 570228)
0 引 言
氮氧化物(NOx)的排放对全球环境及人体健康造成了巨大危害,NH3-SCR技术是当前应用得最为广泛的氮氧化物减排技术之一[1-3]。铁基催化剂以其突出的抗硫性能、优异的热稳定性、良好的中高温催化活性以及价格低廉、绿色环保等优势受到广泛关注[4-5]。然而铁基催化剂仍存在着低温活性差以及温度窗口窄的问题,因此许多研究者以晶相调控或表面结构调变的方法来改善催化剂的氧化还原循环和酸循环,以提升其低温SCR活性并拓宽温度窗口[6-8]。此外,掺杂其他金属氧化物、采用多元复合催化体系改性等方式也能对催化剂进行有效改性。有研究发现向Fe-Ti尖晶石中掺入V/Mn后,NO转化率明显增加,特别是在200~300 ℃时,向Fe-Ti尖晶石加入V对SCR反应的影响比加入Mn明显得多[9]。Zhang等[10]的研究也发现加入少量V形成的铁钒混合氧化物Fe-V-Ox可以提高催化剂的氧化还原性能和催化活性。Mu等[11]制备的钒掺杂Fe2O3催化剂由于氧化还原能力和表面酸性的提高,在175~400 ℃的宽温度范围内可达90%以上的NOx去除率,且同时展现出良好的N2选择性以及抗H2O/SO2性能。近年来,钒酸铁(FeVO4)作为一种具有优异的低温催化活性、宽工作温度窗口以及突出的抗硫性能的金属钒酸盐,受到越来越多研究者的关注。钒酸铁与传统的钒氧物种相比,具有较高的热稳定性[12],因此相较无法规避钒生物毒性的传统VWT催化剂而言,更具绿色环保性。Liu等[13]将钒酸铁负载到不同载体上,发现FeVO4/TiO2催化剂在所有催化剂中具有最佳活性。高分散的FeVO4的活性相只存在于催化剂表面,而并未进入载体TiO2的晶格结构中。且在FeVO4相表面富集的VOx物种是真正的催化活性相,同时催化剂中存在大量与反应物在催化剂表面的吸附和活化有关的表面缺陷。
碱金属及碱土金属化合物(主要为Na2O、K2O、MgO、CaO等)大量存在于以化石、生物质等为燃料的工厂或生活垃圾焚烧过程等排放的烟气中,主要通过对催化剂表面的酸性位点和活性中心的影响来破坏催化反应中的酸循环和氧化还原循环[14-15]。另外,对于金属氧化物基催化剂,碱金属对催化剂的毒害影响一般大于碱土金属,而碱金属中钾的毒害作用又最强[16-17]。然而当前针对铁基催化剂抗碱性能的研究非常少,此外其低温性能也亟待改善。因此设计开发具有低温活性好、抗碱中毒性能佳的新型铁基脱硝催化剂并对其进行相关研究具有重大意义。
本文通过引入V物种制备低温活性优异且温度窗口较宽的氧化钛负载的铁钒催化剂(FeV/TiO2),并通过一系列性能及表征测试研究了V改性对催化剂活性及抗碱性能的影响。
1 实 验
1.1 实验材料
锐钛矿型TiO2、Fe(NO3)3·9H2O、NH4VO3、草酸、氨水,均购于国药集团化学试剂有限公司,去离子水实验室自制。
1.2 催化材料制备
对于负载型铁钒催化剂的制备[13],首先将一定量的草酸超声溶解于50 mL去离子水中形成草酸溶液,向上述草酸溶液中加入一定量的偏钒酸铵并搅拌使之完全溶解,随后再加入一定量的Fe(NO3)3·9H2O(nFe∶nV=1∶1),然后加入锐钛矿型二氧化钛载体。搅拌一段时间后在50 ℃下旋蒸至蒸干。置于100 ℃的烘箱中过夜干燥,将干燥后的产物置于马弗炉中500 ℃下煅烧5 h,将制得的催化剂标记为FeV/TiO2。此外,非负载的铁钒复合氧化物也被用相同的方法制备(除加入载体外),并标记为Fe0.5V0.5Oδ(nFe∶nV=1∶1);同时制备负载型氧化铁催化剂作对比。将一定量的Fe(NO3)3·9H2O溶解于100 mL去离子水中,加入一定量的锐钛矿型二氧化钛载体并在室温下搅拌使之混合均匀,向上述混合溶液中加入适量的氨水,调节pH值至10,室温下连续搅拌3 h,用去离子水过滤洗涤,置于100 ℃的烘箱中过夜干燥后在马弗炉中500 ℃下煅烧5 h,将制得的催化剂记为Fe2O3/TiO2。上述催化剂的负载量均为10%(质量分数)。
碱金属中毒催化剂的制备过程如下:将一定量的硝酸钾溶解于25 mL去离子水中,搅拌使之溶解,将制备的新鲜催化剂加入上述硝酸钾溶液,在50 ℃下旋蒸至蒸干,干燥并煅烧(500 ℃,5 h)后制得钾中毒的催化剂,并将上述催化剂标记为K-Fe2O3/TiO2、K-FeV/TiO2、K-Fe0.5V0.5Oδ,其中浸渍负载上的K2O的质量分数为1%。
1.3 催化材料表征
采用日本Rigaku公司D/Max-RB型X射线衍射(XRD)光谱仪来表征催化剂的晶体结构,扫描范围10°~80°,扫速8(°)·min-1,测试电压40 kV,电流40 mA,λ=0.154 18 nm;采用英国Jobin Yvon公司HR-800型拉曼(Raman)光谱仪测试催化剂表面分子键的振动情况,以532 nm的激光光源作为激发光源;利用美国Micromeritics公司ASAP 2460型氮气物理吸附(BET)仪测试催化剂的比表面积信息,测试前在300 ℃下将样品在脱气系统(VacPrep 061)中预先脱气12 h;使用美国Agilent公司Cary 5000型紫外-可见-近红外(UV-Vis-NIR)分光光度计表征催化剂分子结构及电子转移相关信息,使用BaSO4作为参考,通过漫反射光谱(DRS spectra)记录样品在200~800 nm范围内的吸光度;采用美国Micromeritics公司Auto Chem HP 2950型全自动化学吸附仪进行氨气程序升温脱附(NH3-TPD)及氢气程序升温还原(H2-TPR)实验,以测试催化剂表面酸性及氧化还原性能。具体操作为先称取80 mg催化剂,以10 ℃/min的升温速率在N2气氛下升温至300 ℃保持30 min进行预处理。结束预处理并降温至100 ℃后,通入H2/NH3,进行程序加热升温至900 ℃。
1.4 催化性能测试
催化剂催化活性的测试在固定床反应器中进行(测试所用石英管内径为7 mm)。具体测试条件为:NO、NH3气体浓度为5.0×10-4,O2体积分数为5%,并以N2作为载气,气体总流量设置为260 mL·min-1左右,反应气体体积空速为40 000 h-1。利用FT-IR光谱分析仪(Thermo Fisher Scientific)对进出口反应气NOx、NH3、N2O的浓度进行检测。催化剂的NO转化率和N2选择性通过以下方程计算:
(1)
(2)
2 结果与讨论
2.1 催化剂性能分析
如图1为催化剂的NOx转化率随测试温度变化的曲线。引入V后形成的FeV/TiO2催化剂相较于Fe2O3/TiO2催化剂,其SCR活性显著提升,温度窗口大大拓宽,在210~420 ℃具有90%以上的NOx转化率。对于碱中毒催化剂,K-FeV/TiO2在240~370 ℃温度范围内仍能保持80%以上的催化活性,而K-Fe2O3/TiO2则严重失活,甚至在高温下完全失活。因此,引入V对催化剂的活性及抗碱金属中毒性能具有明显的提升效果。此外,还测试了催化剂的N2选择性(见图2),发现Fe2O3/TiO2催化剂在中毒前后的整个测试温度范围内均能保持95%以上的选择性,FeV/TiO2与之几乎相同但在高温段选择性略有下降,K-FeV /TiO2催化剂在360 ℃以上的高温段下降较为明显。

图1 不同催化剂的NOx转化率Fig.1 NOx conversion over different catalysts

图2 不同催化剂的N2选择性Fig.2 N2 selectivity over different catalysts
2.2 催化剂结构分析

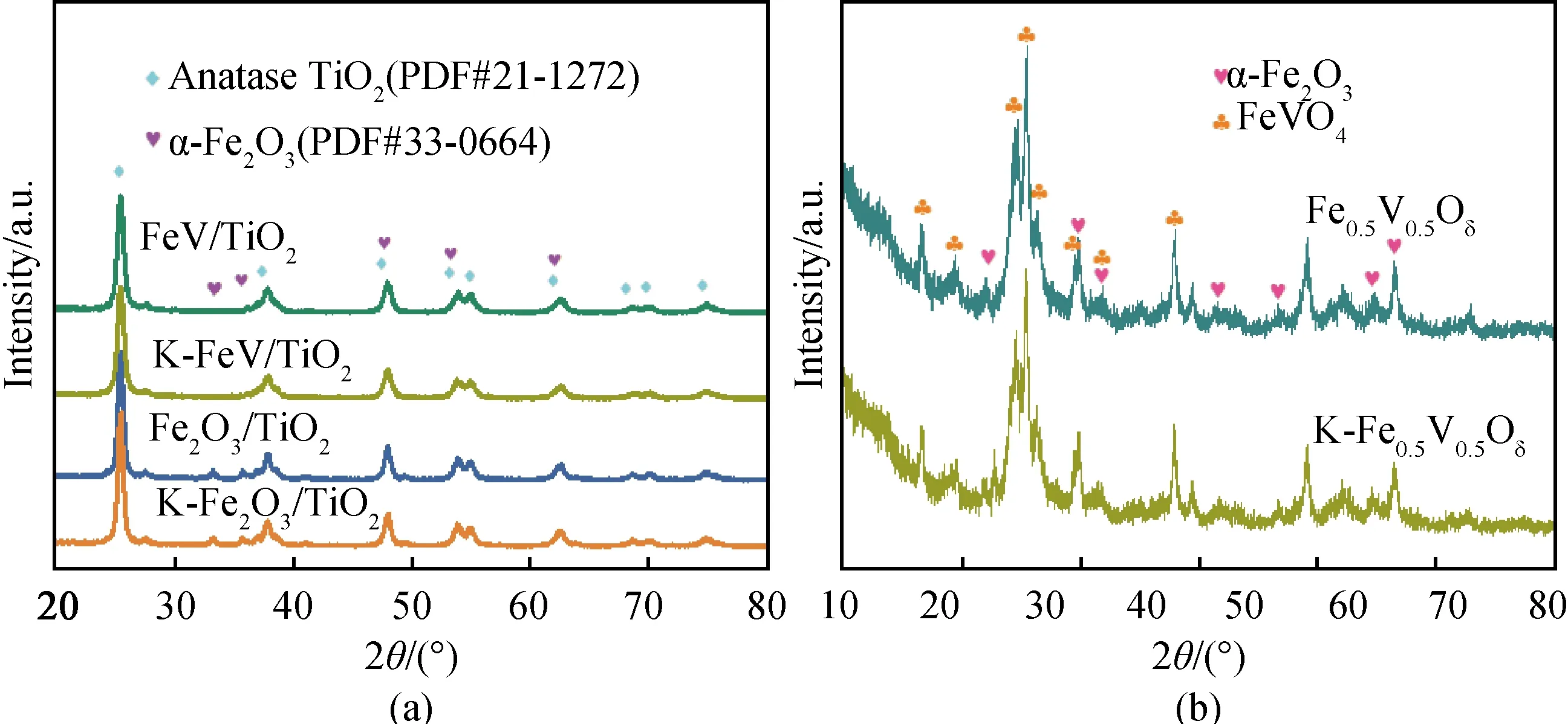
图3 (a)TiO2负载型催化剂和(b)非负载型催化剂的XRD图谱Fig.3 XRD patterns of (a) TiO2 supported catalysts and (b) unsupported catalysts
从Raman图谱可以看到(见图4(a)),出现在143 cm-1、394 cm-1、512 cm-1和635 cm-1处的峰主要是锐钛矿型二氧化钛的特征振动峰[5]。K-Fe2O3/TiO2催化剂中归属于TiO2的Eg峰(143 cm-1)的强度相较于Fe2O3/TiO2的变弱,且发生蓝移(向高波数移动),这说明K可能与TiO2产生了键合作用,导致Ti—O键变得稳定而不易发生断裂,从而不易被活化;而FeV/TiO2催化剂中相应的峰则只是稍许蓝移,并未发生明显变化,说明K中毒对其几乎无影响[15]。同时观察非负载铁钒复合氧化物的Raman图谱(见图4(b))可以发现,非负载Fe0.5V0.5Oδ催化剂的拉曼特征峰主要显示为FeVO4相(320 cm-1、382 cm-1、750 cm-1、822 cm-1以及911 cm-1处)[11,20]。

图4 (a)TiO2负载型催化剂和(b)非负载型催化剂的Raman图谱Fig.4 Raman spectra of (a) TiO2 supported catalysts and (b) unsupported catalysts
此外,由表1中BET测试的结果可以发现,引入V后的FeV/TiO2催化剂相较于Fe2O3/TiO2催化剂,其比表面积显著增大,且在K中毒后仍具有较大的比表面积,从而可为NH3-SCR反应提供更多的活性位点[15]。

表1 不同催化剂的比表面积Table 1 Specific surface area of different catalysts
UV-Vis-NIR的测试结果显示(见图5),Fe2O3/TiO2在547 nm处出现了对应于Fe3+的吸收带[21],而对于FeV/TiO2催化剂,其归属于Fe3+的吸收带出现在512 nm处,较Fe2O3/TiO2发生红移,说明其Fe3+的化学环境由于Fe与V之间强电荷相互作用而发生了明显变化[22]。同时可以观察到,Fe2O3/TiO2在742 nm左右处出现了属于α-Fe2O3的一个典型负带,而在FeV/TiO2催化剂中并未出现,这也说明了后者的分散性更好。此外,这两种催化剂在K中毒后均未发生明显变化。

图5 不同催化剂的UV-Vis-NIR图谱Fig.5 UV-Vis-NIR DRS spectra of different catalysts
2.3 催化剂表面酸性及氧化还原性分析
如图6中的NH3-TPD测试结果所示:与Fe2O3/TiO2相比,FeV/TiO2催化剂的NH3吸附容量大大增加(+55.61%),表明其酸性大大增强;中毒后,相较于K-Fe2O3/TiO2催化剂酸量的大大损失(NH3吸附容量下降了67.42%),FeV/TiO2催化剂在碱中毒后仍能保持相当的酸量,从而保证了NH3-SCR反应中反应气体NH3在催化剂表面的吸附,这也是其在中毒前后均能表现出较高的反应活性的重要原因[23]。

图6 NH3-TPD-MS中不同催化剂的NH3的MS曲线Fig.6 MS signal of NH3 from NH3-TPD-MS profiles for different catalysts
催化剂的氧化还原能力也是影响其催化活性的重要因素。从图7显示的H2-TPR测试结果可知,Fe2O3→Fe3O4、Fe3O4→FeO、FeO→Fe的连续还原过程在Fe2O3/TiO2催化剂中所对应的还原温度分别为324 ℃、363 ℃和426 ℃[5,24]。而在K中毒后分别向高温区域移至381 ℃、418 ℃和514 ℃。在FeV/TiO2中,出现在398 ℃的还原峰为Fe3+→Fe2+与V5+→V4+的共还原峰[25],其后在更高温度出现的还原峰则对应于Fe2+→Fe与V4+→V3+的共还原峰[26]。与Fe2O3/TiO2相比,其还原峰向高温段有所转移,这可能是由于引入V后形成的FeVO4物种抑制了Fe2O3的过度氧化;此外,其尖锐的峰型也说明了引入V后,活性组分在载体上的分散性大幅提高[23]。同时FeV/TiO2及其K中毒的K-FeV/TiO2的还原峰面积较Fe2O3/TiO2及K-Fe2O3/TiO2更大,这说明其H2消耗量较高,也即其氧化还原性显著提升。结合紫外结果及上述对催化剂结构的分析可知,这可能是由于Fe、V之间发生了电荷转移而产生的强电荷相互作用,以及FeVO4与Fe2O3间的电子相互作用[10-11]。
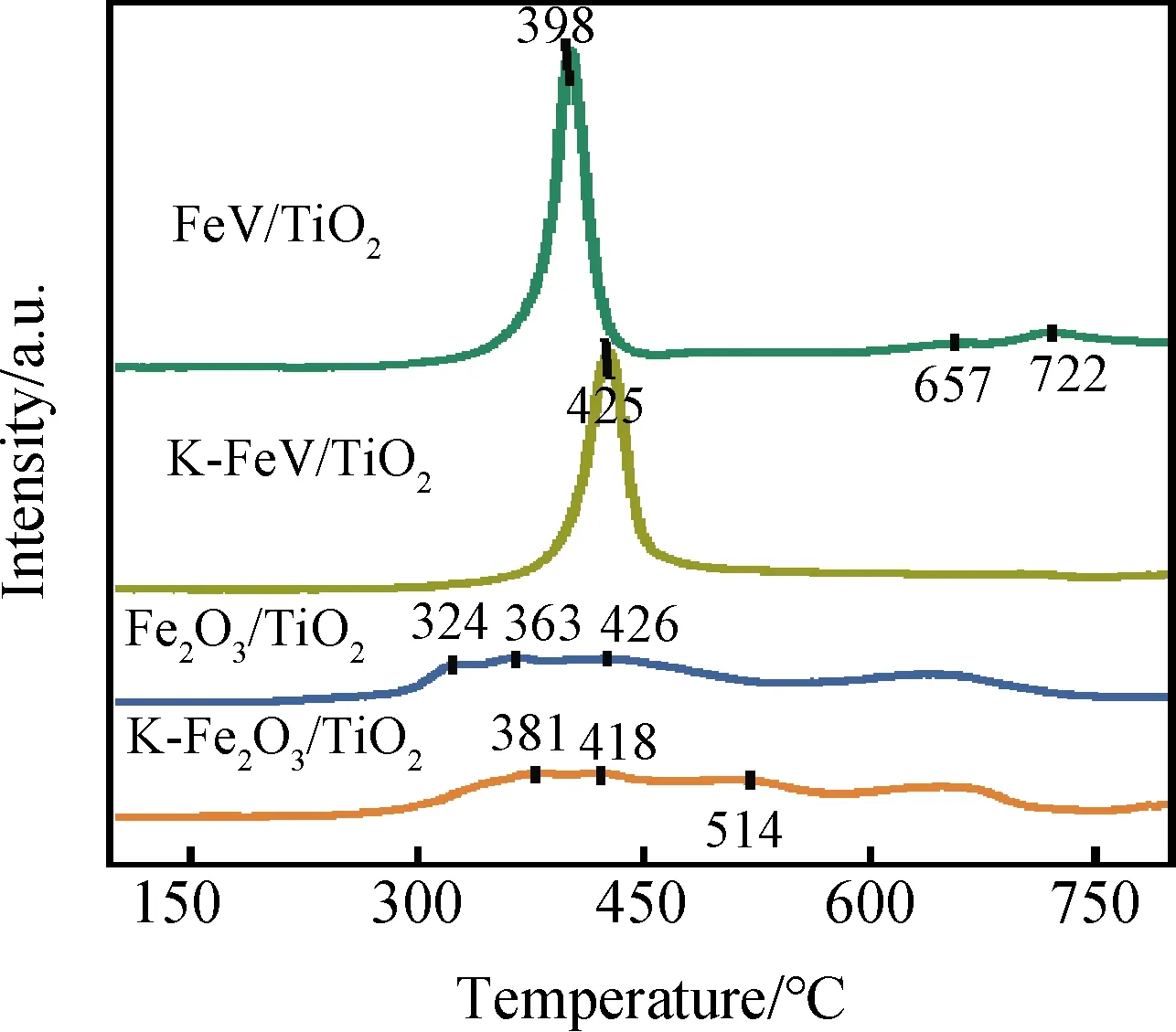
图7 不同催化剂的H2-TPR曲线Fig.7 H2-TPR profiles of different catalysts
3 结 论
通过引入V制备了FeV/TiO2催化剂,与未改性的Fe2O3/TiO2催化剂相比,其低温活性得到显著提升且温度窗口大幅拓宽,碱中毒后,FeV/TiO2催化剂在240~370 ℃内保持80%以上的催化活性。通过对催化剂结构性质和表面理化性质的表征,发现引入V后,催化剂呈现出以FeVO4为主相并伴有少量Fe2O3的状态,且其结构几乎不受K中毒的影响。催化剂中存在大量酸性位点而具有较强的表面酸性,Fe、V物种间的强相互作用也大大提升了其氧化还原性,因此催化剂具有优异的低温活性和较宽的温度窗口。此外,催化剂的结构几乎不受碱中毒的影响,且在K中毒后仍能维持较强的表面酸性,从而使其具有良好的抗碱金属中毒性能。
猜你喜欢
意林原创版(2019年6期)2019-06-30 23:25:04
石油石化绿色低碳(2019年6期)2019-02-13 09:39:01
中学生数理化·八年级物理人教版(2018年6期)2018-06-26 08:36:40
小学生优秀作文(低年级)(2018年3期)2018-02-08 02:20:55
小学生时代(2016年4期)2016-12-12 01:27:17
浙江大学学报(工学版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中国资源综合利用(2016年4期)2016-01-22 08:27:23
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
