
晚发型糖原贮积病Ⅱ型临床、肌肉组织病理学及分子生物学特征分析
2021-09-22 06:28吴世陶刘方石伟伟张敏刘恒方
中国现代神经疾病杂志 2021年6期
吴世陶 刘方 石伟伟 张敏 刘恒方
糖原贮积病Ⅱ型(GSDⅡ),又称Pompe病,是罕见的常染色体隐性遗传性疾病,系GAA基因变异所致[1]。GAA基因变异使酸性α葡糖苷酶(GAA)活性下降或缺失,糖原无法分解利用而沉积于心肌、骨骼肌、平滑肌和肝脏等细胞的溶酶体内,导致一系列临床症状[2]。根据发病年龄和临床表现分为婴儿型(1岁以内发病)和晚发型(1岁以后发病)。晚发型GSDⅡ型主要表现为进行性四肢无力和呼吸困难,易误诊为其他肌肉病,因此,提高早期诊断率十分重要。GSDⅡ型患者具有特征性肌肉组织病理学表现,明确诊断依靠基因检测。近年来,晚发型GSDⅡ型的临床特点和基因变异位点陆续被报道,新发GAA基因变异位点不断被发现,但临床对该病的认识仍严重不足[3]。本研究回顾总结郑州大学第五附属医院诊断与治疗的5例晚发型GSDⅡ型患者的临床、肌肉组织病理学和分子生物学特征,旨在进一步提高认识、减少漏诊和误诊。
临床资料
一、病例选择
1.纳入与排除标准[3]:(1)发病年龄 ≥1岁,临床表现为抬头无力、渐进性对称性四肢近端肌无力,多伴呼吸肌无力,肌张力降低。(2)血清GAA酶活性明显降低。(3)肌肉组织病理学可见肌纤维颗粒-空泡样改变,高碘酸-雪夫(PAS)染色呈阳性。(4)GAA基因检测发现致病性变异。(5)排除线粒体肌病、脂质沉积性肌病和先天性肌营养不良症等疾病。
2.一般资料 选择2013年1月至2020年1月在郑州大学第五附属医院神经内科住院治疗的晚发型GSDⅡ型患者共5例,男性2例,女性3例;发病年龄6~20岁,平均10.82岁;确诊年龄8~31岁,平均15.45岁;病程1~11年,平均4.63年;5例患者均无家族史且父母非近亲婚配。
二、临床特点
5例患者均存在抬头无力、四肢近端肌无力、肌张力降低、不耐受疲劳,3例(例1、例3、例5)呼吸困难,2例(例2、例4)消瘦,2例(例2、例4)腹泻,2例(例3、例5)脊柱侧弯,1例(例4)翼状肩胛和1例(例5)弓形足。3例患者四肢肌力为3~4级,2例(例3、例4)为4级。5例患者血清肌酸激酶(CK,正常参考值36~200 U/L)均轻度升高,为988~1872 U/L、平均1405 U/L;干血滤纸片法检测GAA酶活性[正常参考值1.46~20.34μmol(/L·h)]均明显降低,为0.18~0.37μmol(/L·h)、平均0.27μmol(/L·h)。心电图、心脏和肝脏彩超未见异常;5例患者肌电图均呈肌源性损害;3例(例1、例3、例5)肺功能检查提示限制性通气障碍,多导睡眠图(PSG)监测显示夜间低氧血症。5例晚发型GSDⅡ型患者的临床资料参见表1。
三、肌肉组织病理学特点
1.检测方法 5例患者均行左侧肱二头肌组织活检术,切取新鲜肌肉标本经液氮冷却的异戊烷迅速冷冻,制备7μm层厚组织切片,行HE染色、改良Gomori三色(MGT)、还原型烟酰胺腺嘌呤二核苷酸(NADH)、琥珀酸脱氢酶(SDH)、细胞色素C氧化酶(COX)、油红O(ORO)、PAS和ATP(p H值为4.2和10.6)染色,于光学显微镜下观察组织病理学改变。
2.组织病理学表现 5例患者HE染色均显示肌纤维大小不等,少量变性、坏死的肌纤维散在分布,多数肌纤维内可见大小不一、数量不等、形态不规则的空泡样改变,有4例空泡内有嗜碱性颗粒沉积(图1a)。5例患者MGT染色均可见空泡内大量蓝紫色颗粒沉积(图1b)。1例(例1)PAS染色显示空泡内糖原成分流失(图1c),4例可见多数肌纤维和空泡内糖原成分增多(图1d);5例患者ORO染色未见明显脂滴增多。

图1 晚发型GSDⅡ型患者肌肉组织病理学检查所见 ×200 1a 多数肌纤维内可见大小不一、数量不等、形态不规则的空泡样改变 HE染色 1b 空泡内有大量蓝紫色颗粒沉积 MGT染色 1c 可见空泡内糖原成分流失 PAS染色 1d 可见空泡内糖原成分增多PAS染色Figure 1 The characteristics of muscular pathology of late-onset GSDⅡ ×200 Optical microscopy showed that vacuoles with different sizes and irregular shapes were found in most muscle fibers(Panel 1a).HE staining Optical microscopy showed that a large number of blue and purple particles were deposited in the vacuoles(Panel 1b).MGT staining Optical microscopy showed the loss of glycogen in vacuoles(Panel 1c).PAS staining Optical microscopy showed glycogen shunting in the vacuoles(Panel 1d).PASstaining
四、分子生物学特点
1.检测方法 分别采集5例患者及4例患者(例1、例2、例4、例5)父母的外周静脉血各5 ml,送检北京金准基因科技有限责任公司,提取基因组DNA,打断DNA并制备文库,通过芯片对GSD相关基因编码区及邻近剪切区的DNA进行捕获和富集,采用高通量测序平台行突变位点检测。测序数据剔除接头及低质量数据,采用GATK软件(https://gatk.broadinstitute.org/hc/en-us)分析,找出单核苷酸多态性(SNP)和Indel突变位点;同时与人类基因突变数据库(HGMD,http://www.hgmd.org)及千人基因组数据库(http://browser.1000genomes.org)进行比对,采用SIFT(http://provean.jcvi.org/index.php)、Human Splicing Finder(http://www.umd.be/HSF3/HSF.shtml)、Mutation Taster(http://www.mutationtaster.org/)和Polyphen2(http://genetics.bwh.harvard.edu/pph2/)等多个软件预测变异对蛋白质结构和功能的影响,并参照美国医学遗传学和基因组学会(ACMG)指南进行判断,对可能致病的点突变行Sanger测序验证,并行家系分析。
2.基因检测结果 Sanger测序结果显示,例1及其父存在GAA基因外显子8 c.1320_1322delGAT缺失突变(图2a,2b),导致氨基酸改变p.Met440del(甲硫氨酸缺失),为已知变异;例1及其母存在GAA基因外显子16 c.2331G>C同义突变(图2c,2d),导致氨基酸改变p.Thr777Thr(苏氨酸突变为苏氨酸),HGMD数据库未见收录。例2及其父存在GAA基因外显子16 c.2237G>A无义突变(图3a,3b),导致氨基酸改变p.Trp746*(色氨酸突变为终止),为已知变异;例2及其母存在GAA基因外显子5 c.877G>A错义突变(图3c,3d),导致氨基酸改变p.Gly293Arg(甘氨酸突变为精氨酸),为已知变异。例3存在GAA基因外显子16 c.2238G>C错义突变(图4a)和外显子4 c.784G>A错义突变(图4b),分别导致氨基酸改变p.Trp746Cys(色氨酸突变为半胱氨酸)和p.Glu262Lys(谷氨酸突变为赖氨酸),均为已知变异。例4患者存在GAA基因外显子9 c.1432G>A纯合突变(图5a),导致氨基酸改变p.Gly478Arg(甘氨酸突变为精氨酸);其母存在GAA基因外显子9 c.1432G>A杂合突变(图5b),为携带者,HGMD数据库未见收录;其父该位点未见变异(图5c)。例5及其父存在GAA基因外显子14 c.2014C>T错义突变(图6a,6b),导致氨基酸改变p.Arg672Trp(精氨酸突变为色氨酸),为已知变异;例5及其母存在GAA基因内含子16 c.2332-2A>G剪切突变,导致氨基酸3’端剪切异常(图6c,6d),HGMD数据库未见收录。

图2 例1及其父母Sanger测序结果 2a,2b 例1及其父存在GAA基因外显子8 c.1320_1322delGAT缺失突变(红色圆圈所示)2c,2d 例1及其母存在GAA基因外显子16 c.2331G>C杂合突变(红色圆圈所示)Figure 2 Sanger sequencing results of Case 1 and parents Case 1(Panel 2a)and father(Panel 2b)occurred c.1320_1322delGAT deletion mutation in exon 8 of GAA gene(red circles indicate).Case 1(Panel 2c)and mother(Panel 2d)occurred c.2331G>C heterozygous mutation in exon 16 of GAA gene(red circles indicate).
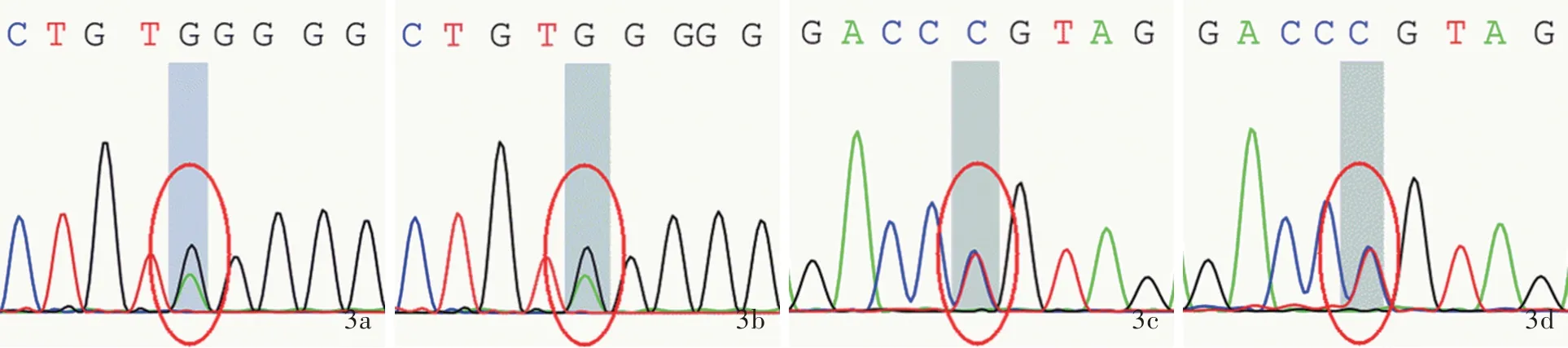
图3 例2及其父母Sanger测序结果 3a,3b 例2及其父存在GAA基因外显子16 c.2237G>A杂合突变(红色圆圈所示) 3c,3d例2及其母存在GAA基因外显子5 c.877G>A杂合突变(红色圆圈所示)Figure 3 Sanger sequencing results of Case 2 and parents Case 2(Panel 3a)and father(Panel 3b)occurred c.2237G>A heterozygous mutation in exon 16 of GAA gene(red circles indicate).Case 2(Panel 3c)and mother(Panel 3d)occurred c.877G>A heterozygous mutation in exon 5 of GAA gene(red circles indicate).
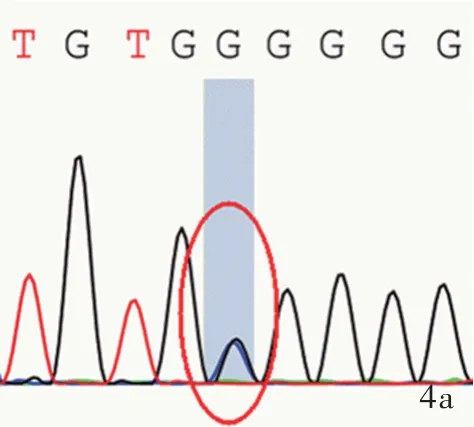
图4 例3患儿Sanger测序结果 4a 例3存在GAA基因外显子16 c.2238G>C杂合突变(红色圆圈所示) 4b 例3存在GAA基因外显子4 c.784G>A杂合突变(红色圆圈所示) 图5 例4及其父母Sanger测序结果 5a,5b 例4及其母存在GAA基因外显子9 c.1432G>A纯合突变(红色圆圈所示) 5c 其父未发生GAA基因变异(红色圆圈所示)Figure 4 Sanger sequencing results of Case 3 Case 3 occurred c.2238G>C heterozygous mutation in exon 16 of GAA gene(red circle indicates,Panel 4a).Case 3 occurred c.784G>A heterozygous mutation in exon 4 of GAA gene(red circle indicates,Panel 4b).Figure 5 Sanger sequencing results of Case 4 and parents Case 4(Panel 5a)and mother(Panel 5b)occurred c.1432G>A homozygous mutation in exon 9 of GAA gene(red circles indicate).Father of Case 4 had no mutation in GAA gene(red circle indicates,Panel 5c).
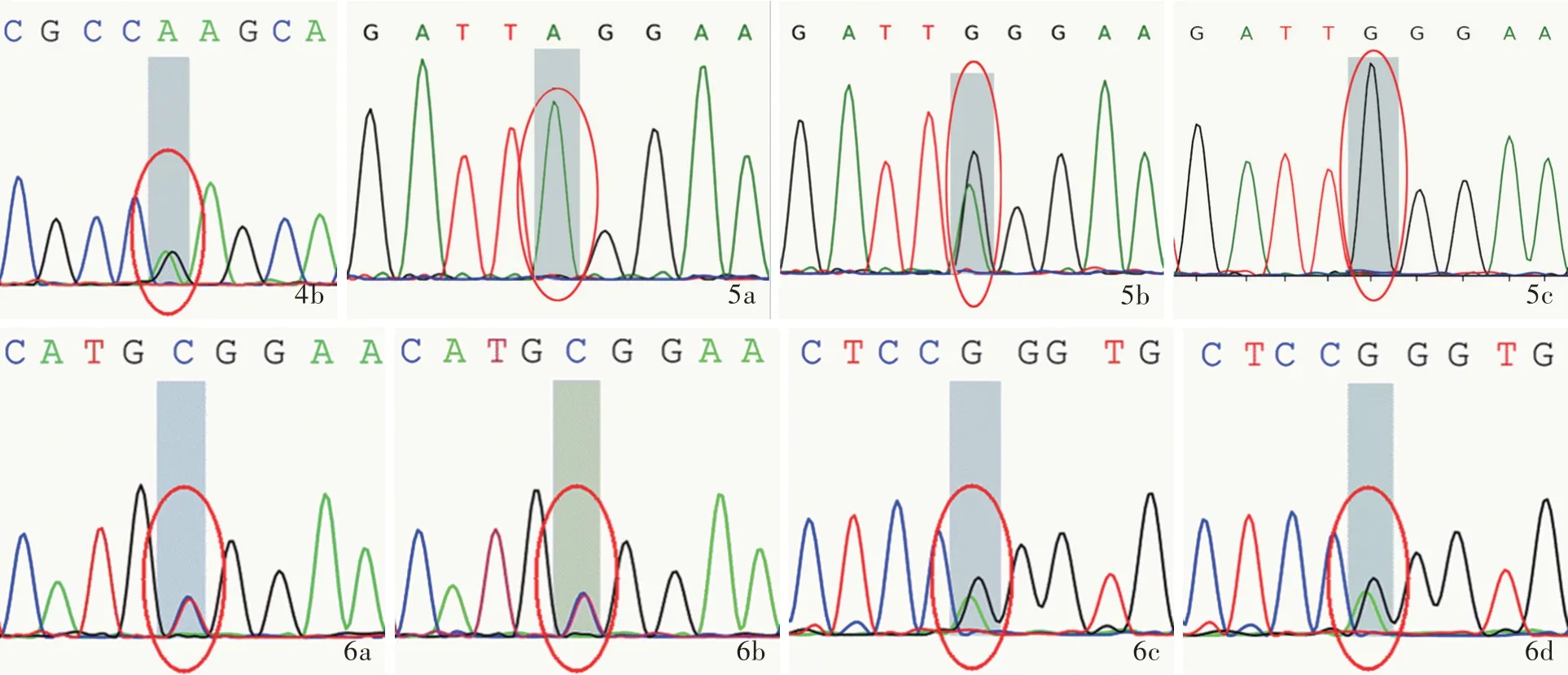
图6 例5及其父母Sanger测序结果 6a,6b 例5及其父存在GAA基因外显子14 c.2014C>T杂合突变(红色圆圈所示) 6c,6d例5及其母存在GAA基因内含子16 c.2332-2A>G剪切突变(红色圆圈所示)Figure 6 Sanger sequencing results of Case 5 and parents Case 5(Panel 6a)and father(Panel 6b)occurred c.2014C>T heterozygous mutation in exon 14 of GAA gene(red circles indicate).Case 5(Panel 6c)and mother(Panel 6d)occurred c.2332-2A>G splicing mutation in intron 16 of GAA gene(red circles indicate).
讨 论
GSDⅡ型的发病年龄、病情严重程度主要与残留的GAA酶活性相关,婴儿型患者GAA酶活性不足正常人的1%,晚发型患者GAA酶活性不足正常人的30%[4]。本组5例患者GAA酶活性均显著下降,临床主要表现为抬头无力、对称性四肢近端肌无力、肌张力降低、不耐受疲劳和呼吸困难,部分患者合并腹泻、消瘦、肌萎缩、脊柱侧弯和弓形足等,与Martínez等[5]的研究结果基本一致。GSDⅡ型患者主要表现为四肢近端肌无力,下肢重于上肢,表现为上下楼梯、跑步和蹲起费力,脑神经支配肌肉较少受累。本组有3例患者存在呼吸困难,肺功能提示限制性通气障碍,多导睡眠图监测显示夜间低氧血症,予无创性呼吸机辅助通气后静脉血氧饱和度正常,提示呼吸肌受累。Momosaki等[6]发现,约50%晚发型GSDⅡ型患者存在呼吸肌和膈肌无力,用力肺活量(FVC)低于正常值的60%,肺部感染诱发呼吸衰竭是致死的主要原因。晚发型GSDⅡ型患者的呼吸肌无力可与肢体无力不平行,部分患者呼吸衰竭前仅有轻至中度肢体无力[7]。尽早检测呼吸功能,及时予无创性呼吸机辅助通气,可以改善晚发型GSDⅡ型患者预后[8]。本组有2例患者自幼腹泻、消瘦,提示胃肠道平滑肌受累。此外,晚发型GSDⅡ型还可出现肌肉疼痛、肌肉痉挛、舌肌肥厚、心肌肥厚和基底动脉瘤等表现[9]。该病之临床表现、血清肌酸激酶水平、肌电图改变与肌肉受累范围及程度相关[10]。本组5例晚发型GSDⅡ型患者血清肌酸激酶水平均轻度升高,与肌纤维变性和坏死相关。GSDⅡ型肌电图多呈肌源性损害,部分患者可出现肌强直放电和复合性重复放电[11],本组5例患者肌电图均呈肌源性损害,未见肌强直放电和复合性重复放电,可能与本组病例较少有关。
GSDⅡ型易误诊为进行性肌营养不良症、先天性肌病、脂质沉积性肌病、线粒体肌病和多发性肌炎等,肌肉组织病理学检测对GSDⅡ型的诊断与鉴别诊断具有重要价值。本组5例患者HE染色显示多数肌纤维内可见大小不一、数量不等、形态不规则的空泡样改变,空泡内沉积嗜碱性颗粒;MGT染色显示空泡内有大量蓝紫色颗粒沉积;4例PAS染色显示多数肌纤维和空泡内糖原成分增多;上述均为GSDⅡ型典型肌肉组织病理改变。此外,1例(例1)可见肌纤维内糖原大量流失,PAS染色呈空泡状,这也是少数GSDⅡ型患者的肌肉组织病理改变特点,应注意与脂质沉积性肌病相鉴别。GSDⅡ型临床表现的严重程度与肌肉组织病理空泡样变性部位和程度相关[12]。部分患者呼吸衰竭前仅有轻度四肢肌无力,肋间肌组织病理学检查显示,约90%的肌纤维发生空泡样变性,而肱二头肌仅约5%的肌纤维发生空泡样变性[13]。GSDⅡ型患者不同取材部位的病理改变差异较大,肌肉病理正常者亦不能排除诊断。此外,线粒体肌病、脂质沉积性肌病和先天性肌病也可见少量糖原沉积,需结合临床资料、多种染色方法和基因检测结果进行鉴别诊断。
GSDⅡ型系GAA基因变异导致的一种罕见的代谢性肌病,呈常染色体隐性遗传,理论上必须为两条等位染色体同时发生致病性变异(复合杂合突变或纯合突变)才可能致病。GAA基因定位于常染色体17q25.2~25.3,相对分子质量为110×103,包含19个内含子和20个外显子,编码952个氨基酸,目前已发现约400余种变异类型[14]。本组例1为c.1320_1322delGAT和c.2331G>C复合杂合突变,其中,c.1320_1322delGAT缺失突变为已知变异[15],而c.2331G>C同义突变未收录在HGMD数据库中,为新发变异,系国内外首次报道,该变异导致氨基酸改变p.Thr777Thr(苏氨酸突变为苏氨酸),蛋白剪切预测软件预测可能影响蛋白剪切而致病,根据ACMG指南评定为致病性变异。本组例4为c.1432G>A纯合突变,导致氨基酸改变p.Gly478Arg(甘氨酸突变为精氨酸),其母亦存在该位点的杂合突变,亦未收录于HGMD数据库中,为新发变异,系国内外首次报道,蛋白结构预测软件预测为致病位点,根据ACMG指南评定为可能致病性变异。本研究例5为c.2014C>T和c.2332-2A>G复合杂合突变,其中,c.2014C>T错义突变为已知变异[16],而c.2332-2A>G剪切突变未收录于HGMD数据库中,该位点人群携带率极低,为新发的剪切突变,系国内外首次报道,该突变导致氨基酸3’端剪切突变,对蛋白功能的影响较大,根据ACMG指南评定为致病性变异。本组5例患者共检出9个GAA基因变异位点,分别位于外显子4、5、8、9、14、16和内含子16,其中3个变异位点位于外显子16,与既往报道的GAA基因变异主要集中于外显子2、10、11和14不同[17-18]。本研究发现c.2331G>C、c.1432G>A和c.2332-2A>G共3个新发变异,扩展了GAA基因变异谱。此外,文献报道亚洲人群GAA基因c.1726G>A和c.2065G>A为假缺陷等位基因,可以降低正常人血清GAA酶活性,但并不致病,不能单独作为诊断GSDⅡ型的依据[19]。目前,基因变异位点与GAA酶活性、临床表型和肌肉组织病理学之间的关系尚不明确,尚待进行大宗病例总结。
综上所述,晚发型GSDⅡ型主要表现为四肢肌无力和呼吸困难,血清GAA酶活性明显下降,肌肉组织病理学具有一定特征性。GAA基因变异主要为复合杂合突变,本研究发现c.2331G>C、c.1432G>A和c.2332-2A>G共3个新发变异,扩展了我国GAA基因变异谱。目前,国内外研究显示GAA酶早期替代治疗可以显著改善预后,基因治疗尚在动物实验阶段[20-21]。本研究有助于加深对晚发型GSDⅡ型的认识,提高早期诊治率,也为产前咨询和产前诊断提供依据。
利益冲突无
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26
中国临床医学影像杂志(2022年5期)2022-07-26
上海交通大学学报(2021年8期)2021-09-02
数字海洋与水下攻防(2021年2期)2021-05-08
种子(2021年3期)2021-04-12
国际放射医学核医学杂志(2021年10期)2021-02-28
水下无人系统学报(2019年1期)2019-03-15
舰船科学技术(2017年11期)2017-11-27
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
