
常染色体隐性遗传性青少年型帕金森病临床特点及疗效评价
2021-09-22 06:28张悦张成李娟吴竞婧林东
中国现代神经疾病杂志 2021年6期
张悦 张成 李娟 吴竞婧 林东
帕金森病是中老年人群常见的神经变性病,临床以静止性震颤、运动迟缓、肌强直和姿势平衡障碍为主要特征,好发于60岁后,40岁前相对少见,平均年龄约55岁。根据发病年龄分为青少年型帕金森病(JOPD,发病年龄<21岁)、早发型帕金森病(EOPD,发病年龄 ≤50岁)和晚发型帕金森病(LOPD,发病年龄 >50岁)[1]。JOPD临床较为少见,呈常染色体隐性遗传者更少见,1973年,Yamamura等[2]首次报告4个常染色体隐性遗传性青少年型帕金森病(AR-JP)家系。JOPD经及时治疗,预后良好,属于可治性遗传病,其中AR-JP对多巴丝肼(美多芭)反应良好,但其引起的症状波动和运动障碍出现早且频繁[1]。本文报道1例AR-JP患者,总结其临床表现、影像学和基因检测结果、治疗与转归,以期提高临床医师对该病的认识,掌握其临床特征、遗传方式、诊断与治疗要点。
临床资料
先证者 女性,27岁,小学教师。主因肢体僵硬、不灵活7年余,四肢不自主抖动5年余,遂于2020年4月8日入院。患者7年前(20岁)开始出现左下肢僵硬、不灵活,行走时向左侧倾斜,当地医院诊断为“运动障碍病因待查”,予中成药治疗(具体方案不详)后效果欠佳,继续予康复理疗后好转,不影响日常生活和学习。5年前(22岁)病情逐渐加重,表现为双下肢僵硬,起步困难,行走缓慢,偶摔倒,并出现四肢不自主抖动,休息可缓解、活动后加重,无意识障碍和精神异常,无肝区疼痛,无饮水呛咳和吞咽困难,无幻觉和性格改变,多次至当地医院就诊,头部和颈腰椎MRI检查均未见异常,予中药和中成药治疗(具体方案不详)后效果欠佳,肢体僵硬缓慢加重。为求进一步明确诊断与治疗,至我院门诊就诊,门诊试验性予以多巴丝肼125 mg/次(3次/d)口服,病情略好转,以“运动障碍病因待查”收入院。患者自发病以来,精神佳,睡眠、饮食可,大小便正常,体重无明显变化。
既往史、个人史及家族史 既往史无特殊,按照国家规定接种疫苗。出生史和生长发育史无特殊。父母非近亲婚配,姊妹共6人,二妹有类似疾病表现(图1)。

图1 AR-JP患者家系图Figure 1 Pedigree of AR-JP.
入院后神经系统查体 神志清醒,言语清晰,对答切题,高级神经功能和脑神经未见异常;双侧瞳孔等大、等圆,直径约2 mm,对光反射灵敏,未见角膜K-F环;四肢不自主抖动,肌力正常,左侧肢体肌张力增高、右侧正常;四肢腱反射活跃,病理反射未引出,共济运动和感觉系统、自主神经系统未见异常,脑膜刺激征阴性。
辅助检查 实验室检查:血尿便常规、血液生化、肝肾功能试验、血清铜蓝蛋白(CP)、凝血功能试验、感染四项[乙型肝炎病毒表面抗原(HbsAg)、丙型肝炎病毒抗体(HCV-Ab)、人类免疫缺陷病毒(HIV)、梅毒螺旋体(TP)抗体]、血管炎四项[核周型抗中性粒细胞胞质抗体(pANCA)、胞质型抗中性粒细胞胞质抗体(c ANCA)、髓过氧化物酶(MPO)、丝氨酸蛋白酶3(PR3)]、风湿四项[类风湿因子(RF)、C-反应蛋白(CRP)、红细胞沉降率(ESR)、抗链球菌溶血素“O”(ASO)]、系统性红斑狼疮六项[抗核抗体(ANA)、抗双链DNA抗体(dsDNA)、抗组蛋白抗体(AHA)、抗核小体抗体(ANuA)、葡萄糖-6-磷酸异构酶(G6PI)、C1q抗体]均于正常值范围,甲状腺球蛋白(TG)抗体1359.80 IU/m(l≤3.99 IU/ml)、甲状腺过氧化物酶(TPO)抗体998 IU/m(l≤9 IU/ml);腰椎穿刺脑脊液常规、生化均于正常值范围,脑脊液查找细菌、隐球菌、抗酸杆菌正常。影像学检查:头部CT未见明显异常;18F-DOPA PET显示,双侧纹状体后部多巴胺代谢降低,提示双侧纹状体多巴胺能神经元功能障碍(图2)。18F-FDG PET显示,全脑葡萄糖代谢未见明显异常(图3)。影像学符合帕金森病代谢改变。
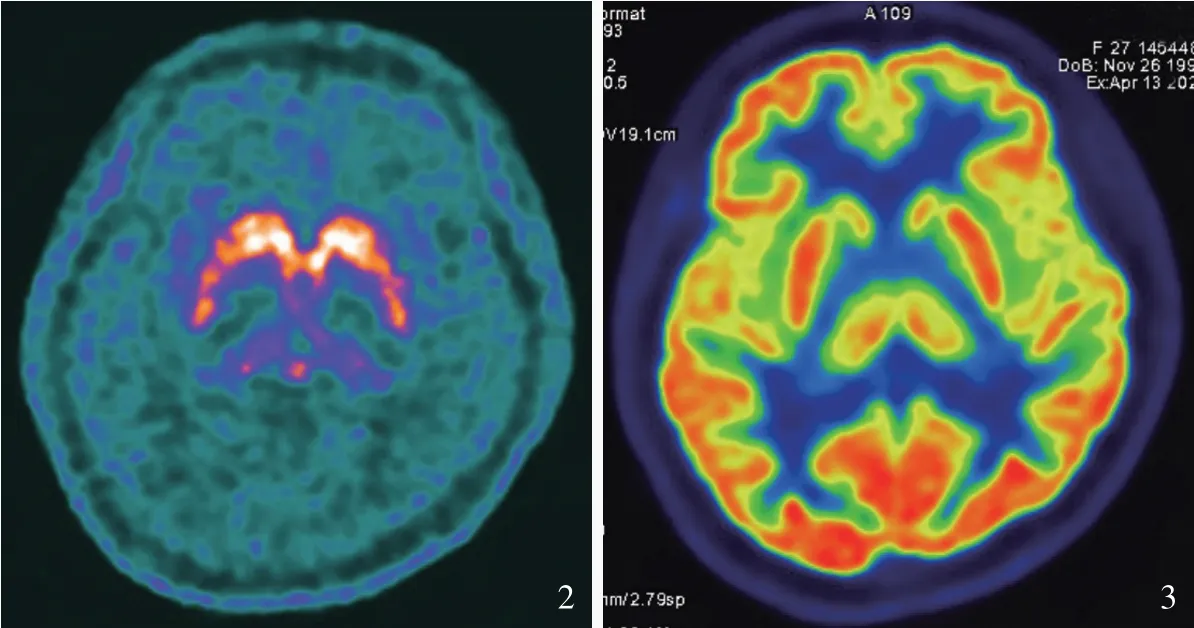
图2 18F-DOPA PET显示,双侧纹状体后部多巴胺代谢降低(紫黄色区域所示)图3 18F-FDG PET显示,全脑葡萄糖代谢正常Figure 2 18F-DOPA PET showed decreased dopamine metabolism in the posterior striatum of both sides(purpleyellow areas indicate).Figure 3 18FFDG PET showed normal glucose metabolism in the whole brain.
基因检测 抽取先证者及其二妹外周静脉血各2~3 ml,送检广州金域医学检验中心,采用多重连接依赖性探针扩增(MLPA)技术检测SNCA、PRKN、UCHLL、PINKI、PARK7、LRRK2、GCH1.ATP13A2基因各外显子是否存在变异,结果显示,先证者存在PRKN基因外显子12 c.1321T>C(p.Cys441Arg)错义突变(图4)以及LRRK2-41(MUT)G2019S和SNCA-2(MUT)A30P杂合缺失(图5)。单独对其二妹PRKN基因变异位点设计引物,行Sanger测序,结果显示其存在PRKN基因外显子12 c.1321T>C(p.Cys441Arg)错义突变。
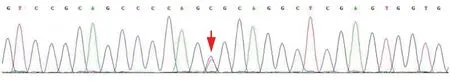
图4 MLPA扩增显示,先证者存在PRKN基因外显子12 c.1321T>C(p.Cys441Arg)错义突变(箭头所示)Figure 4 MLPA amplification showed the proband had a missense mutation in exon 12 c.1321T>C(p.Cys441Arg)of the PRKN gene(arrow indicates).

图5 MLPA扩增结果 5a 可见先证者存在LRRK2-41(MUT)G2019S缺失 5b 可见先证者存在SNCA-2(MUT)A30P缺失Figur e 5 MLPA amplification fingdings The proband had a mutation in LRRK2-41(MUT)G2019S(Panel 5a).The proband had a mutation in SNCA-2(MUT)A30P(Panel 5b).
诊断与治疗经过 先证者在门诊(2020年3月31日)即开始予多巴丝肼125 mg/次(3次/d)口服。综合临床表现、影像学和基因检测结果,最终明确诊断为AR-JP。遂于4月9日调整其治疗方案为多巴丝肼125 mg/次(3次/d)和苯海索(安坦)1 mg/次(3次/d)口服,肢体僵硬、不灵活明显减轻,四肢不自主抖动症状部分减轻;4月14日再次调整为多巴丝肼125 mg/次(3次/d)和苯海索2 mg/次(3次/d)口服,2天后(4月16日)出现异动症,随后改为多巴丝肼125 mg/次(2次/d),同时加服普拉克索(森福罗)125 mg/次(3次/d),但苯海索剂量维持不变;1天后(4月17日)步态僵硬、不灵活及四肢不自主抖动明显减轻,出院。出院后1个月(5月17日)随访,步态僵硬、不灵活及四肢不自主抖动较出院时减轻;2个月后(6月17日)随访,步态僵硬、不灵活明显减轻,四肢不自主抖动基本消失,均未出现异动症和“开关”现象。
讨 论
本文先证者为青少年期发病,起病隐匿,进展缓慢,家族中有类似疾病患者(先证者二妹),临床率先表现为单侧肢体僵硬、不灵活,随后出现动作迟缓,肢体抖动和步态异常出现较晚,且这些症状较其他类型帕金森病轻微,但是病程中未见非运动症状(如自主神经功能障碍、嗅觉减退等),头部PET/CT提示双侧纹状体后部多巴胺代谢降低,对小剂量左旋多巴制剂反应良好,基因检测显示存在PRKN基因变异。综合临床表现、影像学和基因检测结果,最终诊断为AR-JP。
PRKN基因(NM_004562.2)定位于第6号染色体长臂第2区第6条带(6q26),1998年Kitada发现parkin基因,命名为PARK2,该基因在AR-JP中最为常见[3]。针对本文PRKN基因外显子12 c.1321T>C(p.Cys441Arg)错义突变和LRRK2-41(MUT)G2019S和SNCA-2(MUT)A30P杂合缺失,我们做出如下解读:(1)先证者存在PRKN基因2个致病性杂合突变。(2)PRKN基因致病性变异可引起JOPD2型,且常为常染色体隐性遗传模式。(3)PRKN基因外显子12 c.1321T>C突变为错义突变,可使第441位氨基酸由半胱氨酸(Cys)突变为精氨酸(Arg)。该变异未被千人基因组数据库(https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/)和ESP6500siv2 ALL数据库(http://evs.gs.washington.edu/EVS/)收录,而被dbSNP147数据库(www.ncbi.nlm.nih.gov/snp/)收录(rs3765474),且既往有文献报道被检测到[4-6]。进一步行生物信息学分析,错义突变 分 析 采 用PolyPhen2软 件(http://genetics.bwh.harvard.edu/pph2/)、SIFT软 件(http://sift.jcvi.org/)、LRT 软 件 (http://www.genetics.wustl.edu/jflab/lrt_query.html)、MutationTaster软 件(http://www.mutationtaster.org/)、MutationAssessor软 件(http://mutationassessor.org/)、FATHMM软件(http://fathmm.biocompute.org.uk/)、GERP软 件(http://mendel.stanford.edu/SidowLab/downloads/gerp/)、PhyloP软件及 SiPhy 软 件 (https://sites.google.com/site/revelgenomics/),剪接突变分析采用NetGene2 Server软 件(http://www.cbs.dtu.dk/services/NetGene2/)、AUGUSTUS软 件(http://bioinf.uni-greifswald.de/augustus/),认为其致病可能性大。综合考虑,认为该变异为致病性变异。(4)LRRK2-41(MUT)G2019S和SNCA-2(MUT)A30P为杂合缺失突变,可使编码的蛋白质发生大片段缺失,从而丧失正常功能而致病。多篇文献报道,既往在帕金森病患者中检测到该变异[7-10]。
帕金森病的病因和发病机制尚不清楚,认为JOPD与基因和环境因素有关[11]。业已发现,家族性帕金森病与PARK1~13和DJ-1等基因变异相关,但并未发现散发性帕金森病相关基因,考虑与环境因素有关[12]。近年对帕金森病PRKN基因变异的研究较多,累及12个外显子(全部突变占70%,点突变占30%)[13],其中尤以外显子3和4的重新排序以及外显子2和7突变最为常见[14]。原发性帕金森病的发病机制与Zcchc6、Socs7等基因突变有关,为明确诊断和靶基因治疗提供依据[15]。AR-JP的发病机制是PRKN基因变异引起E3酶活性丧失,PRKN蛋白功能障碍而致病[13]。尽管目前有多项关于帕金森病发病机制与基因多态性的研究,但结果不尽一致。尚待大数据对帕金森病及其致病基因的关系进行分析,以期更好了解帕金森病发病机制,有助于诊断、靶基因治疗和预后判断[16]。
18F-FDG PET显像可反映神经细胞葡萄糖代谢水平,神经细胞葡萄糖代谢水平由神经细胞活性和结构完整性决定,帕金森病患者18F-FDG PET表现为结构不完整[17]。18F-DOPA PET显像与神经细胞合成多巴胺的能力有关,帕金森病的发病机制是黑质-纹状体合成多巴胺能力下降,故18F-DOPA PET显像可见黑质-纹状体结构不完整,有助于协助诊断、评估病情以及评价疗效[18-19]。本文先证者头部18F-DOPA PET提示双侧纹状体后部多巴胺代谢降低,考虑纹状体后部突触前神经元多巴胺合成能力下降,引起双侧纹状体多巴胺能神经元功能损害;而18F-FDG PET葡萄糖代谢未见明显异常,考虑双侧基底节区结构正常,提示黑质-纹状体多巴胺能通路变性,符合帕金森病改变,结合病史、体格检查、影像学检查和基因检测,最终确诊为AR-JP。AR-JP早期症状不明显,易误诊,本文先证者误诊长达7年,PET/CT为其诊断AR-JP提供重要依据。
目前尚无相关治疗指南,建议遵循以下治疗原则:(1)左旋多巴制剂是最有效的治疗药物,应从单药小剂量开始,根据病情缓慢加量,以最小剂量达到最佳效果。(2)药物治疗效果欠佳或出现药物导致的运动并发症时,首先考虑药物减量并联合应用苯海索,这是由于大部分患者对苯海索敏感且药物维持时间较长,不良反应较小,若仍需联合用药,可加用普洛卡索[20-21]。(3)药物治疗效果欠佳时,可考虑脑深部电刺激术(DBS),但疗效尚不肯定[21]。大多数患者药物治疗效果良好,早期诊断与及时治疗十分重要,有助于恢复肢体功能、提高生活质量[22]。AR-JP患者早期药物治疗效果良好,但长期治疗部分患者可出现症状波动甚至加重,基因治疗将是新的方向。目前,帕金森病的基因治疗主要通过3条途径:(1)通过基因手段提高脑组织多巴胺水平。(2)通过基因技术减少多巴胺能神经元变性。(3)转染调节基因以减少基因技术的不良反应[23]。研究显示,基因治疗可以改善AR-JP患者临床症状,且无明显不良反应,很可能成为AR-JP治疗的新手段,但目前仍未能在临床推广,主要考虑存在操作困难、治疗效果不稳定、并发症不明确、费用昂贵等原因。
综上所述,本文报道1例20岁发病的AR-JP患者,具有典型临床症状,PET/CT提示双侧纹状体多巴胺能神经元功能损害,基因检测显示PRKN基因复合杂合突变,多巴丝肼治疗效果良好。AR-JP为常染色体隐性遗传性疾病,建议其家族成员完善基因检测,对存在遗传因素的患者,早期诊断、及时治疗。AR-JP系PRKN基因复合杂合突变所致,左旋多巴制剂治疗有效,与苯海索和普拉克索联合应用效果更佳,可减少异动症和“开关”现象,基因治疗可能是治疗方面的新突破。
利益冲突无
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
临床输血与检验(2022年3期)2022-06-22
实用肝脏病杂志(2022年2期)2022-03-21
中国体育科技(2022年1期)2022-03-10
中国康复(2021年6期)2021-11-30
青岛大学学报(医学版)(2021年5期)2021-11-17
南京医科大学学报(自然科学版)(2021年8期)2021-10-19
中国生殖健康(2020年4期)2021-01-18
诊断学(理论与实践)(2020年1期)2020-04-28
首都体育学院学报(2019年5期)2019-10-18
