
骨骼肌磁共振成像在遗传性肌肉病诊断中的应用进展
2021-09-22 06:28宋佳庞咪李刚付俊马明明
中国现代神经疾病杂志 2021年6期
宋佳 庞咪 李刚 付俊 马明明
骨骼肌MRI被认为是检测肌肉病变最敏感、最特异的成像技术,具有软组织分辨率高、可重复、无辐射、不受配合度和疾病严重程度影响等优点,近年越来越多地应用于肌肉病的诊断、病情评估、组织活检定位和追踪随访[1-3]。肌肉病根据病因分为获得性和遗传性两大类,前者主要包括炎性肌病和内分泌性肌病,后者主要包括肌营养不良症、远端型肌病、先天性肌病(CMs)、代谢性肌病和离子通道病等,这些肌病的治疗和预后完全不同,因此,早期准确诊断是必要的。影像学主要表现为信号强度改变(脂肪浸润和水肿)以及肌肉大小或形状改变(肌萎缩、肥大和假肥大),主要集中在下肢MRI,扫描序列包括T1WI和短时间反转恢复(STIR)。脂肪浸润在T1WI上呈高信号,可反映慢性期病变[4]。参照改良Mercuri评分[5],根据T1WI显示的肌肉受累范围和程度评价脂肪浸润程度:0为肌肉信号正常;1为肌肉内散在斑点状高信号;2为肌肉内片状高信号,病灶体积<肌容积的30%;3为肌肉内高信号病灶占肌容积的30%~60%;4为肌肉内高信号病灶>肌容积的60%;5为终末期改变,肌肉组织被脂肪组织或纤维结缔组织替代,呈弥漫性高信号。STIR序列水肿呈高信号、脂肪组织呈低信号,由于炎性肌病的主要改变是水肿,以及多种肌营养不良症肌肉水肿早于脂肪替代,故STIR序列高信号/水肿可以反映炎症和病变活动期。由此可见,MRI通过观察骨骼肌形态和信号改变以显示肌肉受累范围和程度,业已成为诊断肌肉病的重要方法,为临床试验和纵向研究提供了一种有前途的研究工具[6]。本文拟就遗传性肌肉病骨骼肌MRI显示的肌肉选择性受累模式进行综述,以提高神经内科医师对骨骼肌MRI在肌肉病诊断中独特价值的认识。
一、肌营养不良症
肌营养不良症是一组临床异质性、缓慢进展的致残性肌肉病,主要表现为骨盆带和肩胛带肌无力,包括假肥大型肌营养不良症、强直性肌营养不良症(DM)、面-肩-肱型肌营养不良症(FSHD)和肢带型肌营养不良症(LGMD)等。MRI主要表现为受累肌肉脂肪替代信号伴或不伴水肿信号,不同类型易受累部位不同[6]。
1.假肥大型肌营养不良症 亦称为抗肌萎缩蛋白病,系dystrophin基因突变导致的X连锁隐性遗传性疾病,临床异质性较大,主要有症状较严重的Duchenne型肌营养不良症(DMD)和症状较轻微的Becker型肌营养不良症(BMD)[7]。DMD是最常见、最严重的肌营养不良症,发病率为1/3500活产男婴,以骨骼肌细胞胞膜Dystrophin蛋白表达缺失为特征,导致肌肉慢性进行性破坏,从而被脂肪组织和纤维结缔组织替代,通常于3~5岁发病,临床主要表现为进行性加重的四肢近端肌无力伴腓肠肌肥大[8],于13岁左右丧失行走能力,并于成年早期死于呼吸衰竭[9]。肌细胞坏死自大腿开始,随着时间推移,肌肉组织逐渐被脂肪组织和纤维结缔组织替代。与DMD的Dystrophin蛋白完全缺失不同,BMD尚存部分有功能的Dystrophin蛋白,通常发病较晚、症状较轻、进展较慢[6]。MRI可用于评估肌萎缩和脂肪替代程度,包括亚临床阶段,由于脂肪替代或肌萎缩在疾病早期阶段尚不显著,STIR序列可早期发现肌肉水肿,对于年幼患儿大有裨益[10]。DMD患者根据其肌肉脂肪浸润程度表现出独特的肌肉受累模式:疾病早期可无脂肪浸润,通常发生于6~7岁,起初臀大肌和大收肌脂肪浸润明显,随后为股四头肌和股二头肌,而股薄肌、缝匠肌、长收肌和半腱肌选择性保留,呈“三叶草挂果征”[11-12];腓肠肌较小腿其他肌肉脂肪浸润更加严重;有趣的是,未发生脂肪浸润的肌肉STIR序列呈高信号,提示肌肉水肿可能早于脂肪替代[9]。BMD的骨骼肌MRI显示的肌肉受累模式与DMD相类似,但病变程度较轻微[12](图1)。

图1 男性BMD患者,17岁。骨骼肌MRI检查所见 1a 横断面T1WI显示,双侧大腿缝匠肌、股薄肌、半腱肌和长收肌脂肪浸润相对较轻,其余肌肉均为重度脂肪浸润 1b 横断面T1WI显示,双侧小腿腓肠肌和腓骨肌脂肪浸润较明显Figure 1 A BMD male patient with 17-year-old,lower limb muscles MRIfindings Axial T1WIshowed severe fatty degeneration of almost all thigh muscles with relative sparing of the sartorius,gracilis,semitendinosus and adductor longus muscles(Panel 1a).Axial T1WI showed obvious fatty degeneration of bilateral gastrocnemius and peroneus muscles in the calves(Panel 1b).
2.强直性肌营养不良症 系第2位常见的肌营养不良症,亦是成人最常见的肌营养不良症[13]。根据致病基因分为强直性肌营养不良症1型和2型(DM1和DM2)。DM1型的致病基因定位于染色体19q13.3,系由DMPK基因3’非翻译区(3'UTR)胞嘧啶-胸腺嘧啶-鸟嘌呤(CTG)异常重复扩增所导致;DM2型系定位于第3号染色体的CNBP基因(亦称ZNF9基因)内含子1胞嘧啶-胞嘧啶-胸腺嘧啶-鸟嘌呤(CCTG)异常重复扩增所致[14]。骨骼肌MRI显示,DM1型患者下肢远端肌群脂肪浸润程度较近端严重,其中,小腿脂肪浸润最严重的是腓肠肌内侧头,其次为比目鱼肌和胫骨前肌,而胫骨后肌则相对保留[15];大腿前部肌群脂肪浸润较后部肌群显著,尤以股中间肌和股内侧肌最为严重,而股直肌相对保留[13];多数患者还可同时出现肌肉水肿[15](图2)。DM2型患者的肌肉病变程度较DM1型轻微,大多数DM2型患者下肢骨骼肌MRI无明显异常,但仍有一些患者可出现脂肪浸润,其中,大腿后部肌群较其他肌群易受累,小腿各肌群受累程度相类似[16]。
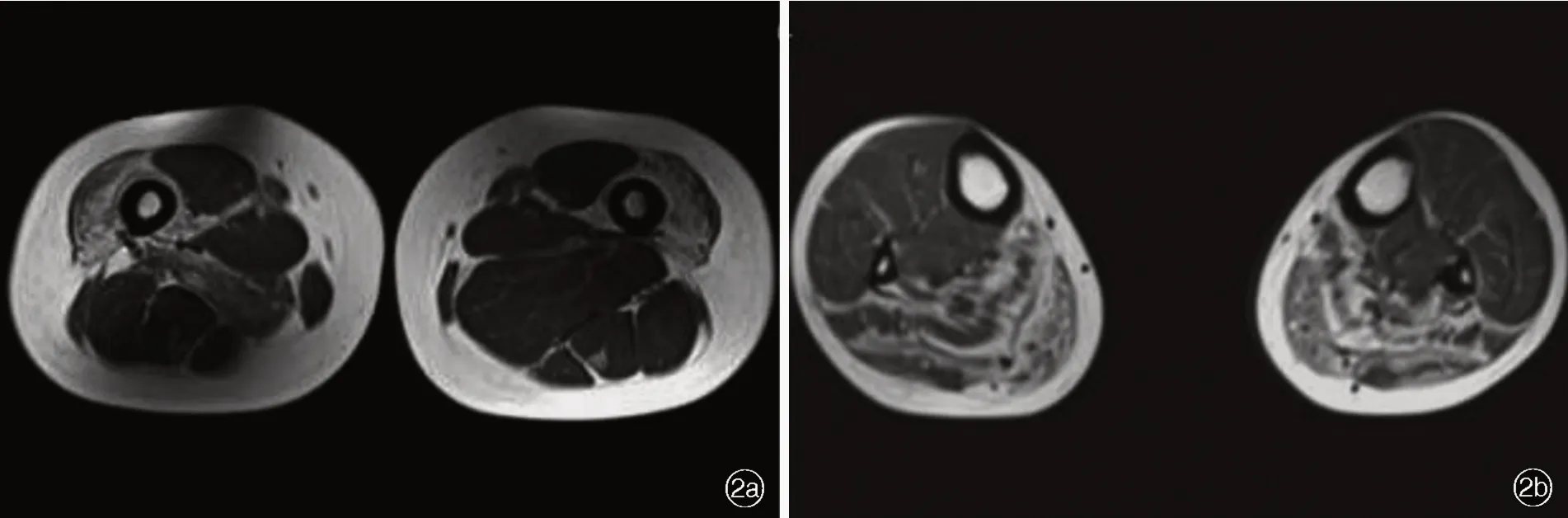
图2 女性DM1型患者,47岁。骨骼肌MRI检查所见 2a 横断面T1WI显示,双侧大腿前部肌群(除外股直肌)和右侧大收肌明显脂肪浸润 2b 横断面T1WI显示,双侧小腿后部肌群尤其是比目鱼肌和腓肠肌内侧头和外侧头明显脂肪浸润Figure 2 A DM1 female patient with 47-year-old,lower limb muscles MRIfindings Axial T1WIshowed severe fatty degeneration of the right adductor magnus and bilateral anterior thigh compartments except the rectus femoris(Panel 2a).Axial T1WI showed bilateral soleus and gastrocnemius affected severely in the calves(Panel 2b).
3.面-肩-肱型肌营养不良症 系第3位常见的肌营养不良症。肌肉不对称性受累是其特征性MRI表现,有文献报道,78%~80%的FSHD患者呈现肌肉不对称性受累[17-18],大腿受累最严重的肌肉为股后肌群和股内收肌[17,19];小腿受累轻微或不受累,最先累及比目鱼肌、腓肠肌内侧头和胫骨前肌,而腓骨肌和胫骨后肌保留[20](图3)。肌肉水肿是常见MRI表现[17]。肌肉组织活检和骨骼肌MRI显示,STIR序列呈高信号的FSHD患者均存在肌肉组织炎症性病理改变以及炎症反应过程中相关基因表达上调,表明肌肉水肿可反映炎症性改变[21]。Beevor征见于腹直肌下半部无力而上半部肌力正常的情况,即患者仰卧位用力抬起头部,检查者以手压住患者头部可见其脐孔向上移动,多见于胸髓病变,亦见于FSHD、GNE肌病、糖原贮积病Ⅱ型(GSDⅡ,亦称Pompe病)和散发性包涵体肌炎(sIBM)[6,22]。

图3 女性FSHD患者,38岁。骨骼肌MRI检查所见 3a 横断面T1WI显示,右侧大腿股后肌群、股中间肌、股内侧肌、股薄肌和双侧大收肌脂肪浸润最严重,其次为右侧股外侧肌和左侧股后肌群 3b 横断面T1WI显示,仅右侧小腿腓肠肌内侧头重度脂肪浸润Figure 3 A FSHD female patient with 38-year-old,lower limb muscles MRI findings Axial T1WI showed the right hamstrings,vastusintermedius,vastusmedialis,gracilis and bilateral adductor magnus muscles affected most severely in the thighs,followed by right vastuslateralisand left hamstrings(Panel 3a).Axial T1WIshowed severefatty degeneration of right gastrocnemiusmedialisin the calves(Panel 3b).
4.肢带型肌营养不良症 系一组临床异质性的常染色体显性或隐性遗传性肌肉病,以缓慢进行性加重的四肢近端肌无力和肌萎缩为特点。肢带型肌营养不良症2A型(LGMD2A,calpain基因变异所致)[23]、2B型(LGMD2B,dysferlin基因变异所致)[24]是最常见的常染色体隐性遗传性亚型,其次为LGMD2C型、LGMD2D型、LGMD2E型、LGMD2F型(分别由编码肌聚糖γ、α、β、δ4个亚单位基因变异所致)[25]、LGMD2I型(FKRP基因变异所致)[26]。明确诊断主要依靠基因检测,而骨骼肌MRI可以指导基因检测。Tomas等[6]报告BMD与LGMD在MRI上的差异表现:股四头肌在BMD患者中受累而在LGMD患者中相对保留。LGMD2A型以大腿后部肌群和内侧肌群受累明显,股四头肌受累相对轻微;小腿以腓肠肌内侧头受累最严重,胫骨后肌相对保留[23](图4)。LGMD2B型大腿脂肪浸润无特异性,所有肌群均可受累,缝匠肌和股薄肌相对保留;小腿受累最严重的是比目鱼肌,其次为腓肠肌,至疾病晚期所有小腿肌群均可受累,包括胫骨后肌[24,27](图5)。LGMD2C~2F型统称为肌聚糖病,其分别由编码肌聚糖γ、α、β、δ4个亚单位的基因变异所致[25]。肌聚糖病共同的脂肪受累模式为,受累最早和最严重的大腿肌肉是大收肌,其次是长收肌、股二头肌长头、半膜肌和股中间肌,大腿后部肌群中半膜肌常较半腱肌受累严重,大腿前部肌群中股中间肌受累最严重、其次是股内侧肌内侧部,股外侧肌远端相对保留,即使至疾病晚期,缝匠肌和股薄肌相对保留或受累轻微;小腿肌群相对保留或仅轻微 受 累 ,尤 以 小 腿 前 部 肌 群 受 累 较 明 显[4,28]。LGMD2I型臀大肌受累显著,其次是大收肌和股二头肌,大腿前部肌群中股中间肌和股外侧肌较股直肌和股内侧肌受累明显[26];小腿后部肌群较前部肌群受累明显,但并无LGMD2A型腓肠肌内头较外头受累显著的差异[29]。
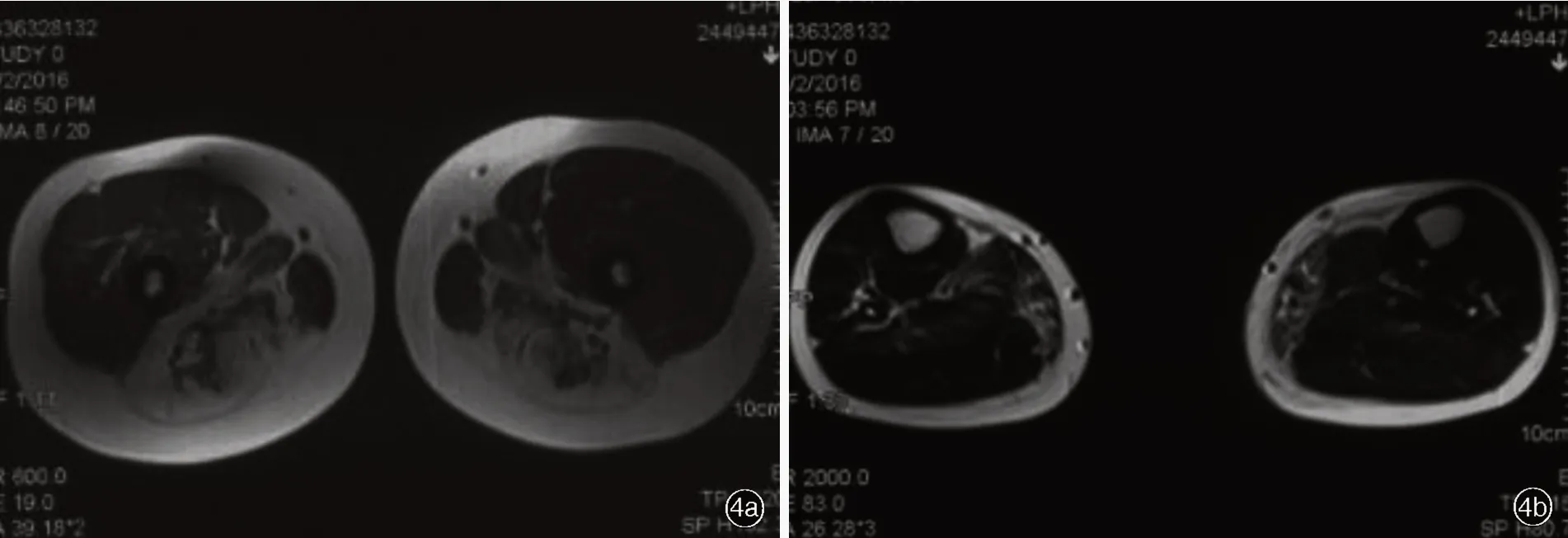
图4 男性LGMD2A型患者,31岁。骨骼肌MRI检查所见 4a 横断面T1WI显示,双侧大腿股后肌群和大收肌重度脂肪浸润4b 横断面T1WI显示,双侧小腿腓肠肌内侧头脂肪浸润最严重Figure 4 A LGMD2A male patient with 31-year-old,lower limb muscles MRIfindings Axial T1WIshowed severe fatty degeneration of bilateral hamstring and adductor magnus muscles(Panel 4a).Axial T1WIshowed obvious fatty degeneration of bilateral gastrocnemius medialis in the calves(Panel 4b).

图5 女性LGMD2B型患者,46岁。骨骼肌MRI所见 5a 横断面T1WI显示,双侧大腿股薄肌、长收肌和缝匠肌脂肪浸润相对较轻微,余肌肉弥漫性重度脂肪浸润 5b 横断面T1WI显示,双侧小腿比目鱼肌重度脂肪浸润,左侧腓肠肌内侧头片状脂肪浸润Figure 5 A LGMD2B female patient with 46-year-old,lower limb muscles MRI findings Axial T1WI showed severe fatty degeneration of almost all thigh muscles with relative sparing of the gracilis,adductor longus and sartorius(Panel 5a).Axial T1WI showed severe fatty degeneration of the soleus,and flaky fatty degeneration of left gastrocnemius medialis in the calves(Panel 5b).
5.胶原蛋白Ⅵ相关肌病 系常染色体显性遗传性疾病,主要包括Bethlem肌病和临床更严重的Ullrich先天性肌营养不良症,与胶原蛋白Ⅵ基因变异相关,临床以缓慢进展性肌无力和关节挛缩为特征性表现,关节挛缩主要影响手指和肘部,还可出现脊柱僵直。MRI常有特征性表现,包括股外侧肌外缘脂肪浸润而肌肉中心相对保留(“三明治征”)以及股直肌中央脂肪浸润(“靶征”)[4,30-31]。
二、远端型肌病
Miyoshi肌病(MM)和GNE肌病是最常见的远端型肌病。(1)Miyoshi肌病:以下肢远端后部肌群受累起病,与LGMD2B型的致病基因一致,均为DYSF基因变异导致其编码的Dysferlin蛋白结构或功能异常引起的不同临床亚型,与LGMD2B型肌肉受累模式相似[24]。(2)GNE肌病:系GNE基因变异引起的临床少见的常染色体隐性遗传性疾病,特征性骨骼肌病理改变为肌纤维内镶边空泡形成,亦称伴镶边空泡的远端型肌病(DMRV),影像学特点为下肢远端肌群受累显著,小腿以胫骨前肌和比目鱼肌受累最严重,腓肠肌特别是腓肠肌外侧头受累最轻微,但随着疾病进展,所有小腿肌群均可受累;大腿以后部肌群和内侧肌群受累最严重,股四头肌相对保留,尤其是股外侧肌和股直肌即使至疾病晚期也相对保留[32-33](图6)。

图6 男性GNE肌病患者,34岁。骨骼肌MRI检查所见 6a 横断面T1WI显示,双侧大腿股外侧肌和股直肌相对保留,其余肌肉重度脂肪浸润 6b 横断面T1WI显示,双侧小腿肌肉均重度脂肪浸润Figur e 6 A GNE myopathy male patient with 34-years-old,lower limb muscles MRI findings Axial T1WI showed severe fatty degeneration of almost all thigh muscles except vastuslateralis and rectus femoris(Panel 6a).Axial T1WIshowed severe fatty degeneration of all lower leg muscles(Panel 6b).
三、先天性肌病
先天性肌病是罕见的常染色体显性或隐性遗传性肌病,具有临床异质性,根据骨骼肌组织病理学表现分类,分为中心核肌病(centronuclear myopathy)、杆状体肌病(nemaline myopathy)、中央轴空病(central core disease)、多微小轴空病(MmD)和先天性肌型比例失调(congenital fiber type disproportion)[34]。先天性肌病患者头面、骨盆带和肢体肌肉可发生脂肪替代,不同亚型的脂肪替代模式不同[6]。(1)中心核肌病:特征性骨骼肌MRI表现为,除股直肌外的股四头肌、缝匠肌和大收肌受累显著,股直肌、长收肌和股后肌群相对保留;小腿比目鱼肌、腓肠肌外侧头和腓骨肌受累显著,小腿前部肌群和腓肠肌内侧头相对保留[29]。(2)杆状体肌病:骨骼肌MRI表现具有高度异质性,并与临床表型高度异质性和不同基因变异类型相对应[29]。NEB基因变异是引起杆状体肌病的最常见原因;ACTA1基因变异是第2位常见原因,且大多数患者的临床表型严重[35]。有文献报道,NEB基因变异致杆状体肌病以下肢远端肌萎缩为主,而ACTA1基因变异致杆状体肌病与下肢近端肌肉受累相关;而且在NEB基因变异致杆状体肌病患者中存在一种独特的肌肉受累模式,较少或不累及大腿肌群,但选择性累及股直肌[35-36],疾病早期小腿胫骨前肌受累,选择性累及小腿前部肌群的模式与中心核肌病相反,后者胫骨前肌通常不受累;ACTA1基因变异致杆状体肌病患者大腿缝匠肌和大收肌受累最严重,股二头肌、半腱肌和半膜肌均匀受累,小腿主要累及胫骨前肌和腓骨长肌[37]。此外,晚发型或成人型杆状体肌病几乎未发现致病性突变,可能与单克隆免疫球蛋白病或人类免疫缺陷病毒(HIV)感染有关,特别是晚发型散发性杆状体肌病患者最常累及大腿后部肌群(尤其是半膜肌),而选择性累及小腿比目鱼肌[38]。(3)多微小轴空病:系临床高度异质性肌病,selenoprotein N基因突变引起的经典型多微小轴空病选择性累及缝匠肌[29]。目前关于先天性肌病的MRI研究较少,相同的病理改变可由不同基因变异引起,同一致病基因也可引起不同病理改变,且临床异质性较高、发病率较低,因此其肌肉受累模式尚待进一步探讨。
四、代谢性肌病
代谢性肌病包括线粒体病(MD)、脂质沉积性肌病(LSM)和糖原贮积病(GSD)。目前骨骼肌MRI尚未广泛应用于代谢性肌病,主要集中于GSDⅡ型。GSDⅡ型是临床罕见的常染色体隐性遗传性肌病,由编码溶酶体酸性α葡糖苷酶(GAA)的GAA基因突变引起GAA蛋白缺乏,从而导致糖原在多种组织包括骨骼肌、心肌和平滑肌细胞溶酶体内沉积,阿糖苷酶α替代治疗是目前唯一被中国国家食品药品监督管理总局(CFDA)批准的方法。GSDⅡ型患者骨骼肌MRI显示,最易发生脂肪替代的下肢近端肌肉为大收肌、大腿后部肌群(尤其是半膜肌)和股中间肌,随着疾病进展,股外侧肌和股内侧肌受累,而股直肌、缝匠肌、股薄肌和股外侧肌浅层相对保留,下肢远端肌肉通常不受累或轻微受累[6,39](图7)。核黄素反应性脂质沉积性肌病(RR-LSM)是我国最常见的脂质沉积性肌病,骨骼肌MRI表现为轻度脂肪浸润,尤以大腿后部肌群和大收肌明显[40-41](图8)。中性脂质沉积病(NLSD)是罕见的遗传性脂肪代谢异常疾病,系脂滴在不同组织沉积,临床以成人发病的进行性肌肉病为主要特征,与心肌病、肝脂肪变性和身材矮小等有关,肌无力呈弥漫性,通常发生于上肢近端和下肢远端。骨骼肌MRI显示,下肢比目鱼肌、腓肠肌内侧头、臀小肌和半膜肌受累最严重,其次为臀中肌、股二头肌长头、长收肌、大收肌、胫骨后肌和胫骨前肌[42]。应注意的是,病程早期即累及胫骨后肌,与大多数肌肉病特别是LGMD和远端型肌病相反,后两者即使至疾病晚期胫骨后肌也通常不受累;大腿股四头肌在疾病后期受累,但较大腿后部肌群受累轻微,至疾病晚期可累及除股薄肌和缝匠肌外的下肢所有肌肉;此外,约50%中性脂质沉积病患者STIR序列呈高信号[42]。

图7 男性GSDⅡ型患儿,13岁。骨骼肌MRI检查所见 7a 横断面T1WI显示,双侧大腿仅大收肌轻度脂肪浸润 7b 横断面T1WI显示,双侧小腿仅胫骨前肌轻度脂肪浸润Figure 7 A GSDⅡmale patient with 13-year-old,lower limb muscles MRIfindings Axial T1WIshowed mild fatty degeneration of bilater adductor magnus mucles in the thighs(Panel 7a).Axial T1WIshowed mild fatty degeneration of bilateral tibialis anterior muscles in the calves(Panel 7b).

图8 男性RR-LSM患者,20岁。骨骼肌MRI检查所见 8a 横断面T1WI显示,双侧大腿部分肌肉有斑点状脂肪浸润 8b 横断面T1WI显示,双侧小腿部分肌肉有斑点状脂肪浸润Figure 8 A RR-LSM male patient with 20-year-old,lower limb muscles MRIfindings Axial T1WIshowed spot-speckled fatty degeneration in some thigh muscles(Panel 8a).Axial T1WIshowed spot-speckled fatty degeneration of some lower leg muscles(Panel 8b).
五、离子通道病
离子通道病主要包括先天性肌强直(氯离子通道变异)、先天性副肌强直(钠离子通道变异)和周期性麻痹(钙离子、钠离子和钾离子通道变异均可),通常MRI无异常[6]。
骨骼肌MRI除上述根据肌肉受累模式提供诊断线索外,还可以指导肌肉组织活检,后者是明确诊断的必要手段,选择适宜的活检部位可以提高阳性检出率,避免重复活检,从而减少诊断与治疗的延误。此外,骨骼肌MRI还可以监测疾病进展和治疗反应,在涉及神经肌肉病的临床试验中,功能检测如6分钟步行试验(6MWT)和运动功能评价量表(MFM)常作为临床试验的终点,功能检测依赖患者的表现与合作,故在儿童和不能行走的患者中是无法开展的。骨骼肌MRI检查几乎不依赖患者的合作,对疾病进展的监测更敏感。例如,DMD患者肌肉脂肪含量随访3个月后即显著变化,随访1年后即使6MWT评分改善或保持稳定者,肌肉脂肪含量仍增加;LGMD2I型患者用力肺活量(FVC)随访1年后下降,尽管MRI提示大部分下肢肌肉脂肪浸润进展明显,而肌力、上下楼梯、坐位站起,以及10米步行试验(10MWT)和6MWT评分均保持不变[4]。
综上所述,骨骼肌MRI可以清晰评估肌肉病变,结合临床症状与体征,可指向单一诊断或有限鉴别诊断,即骨骼肌MRI可以为肌肉病诊断提供依据,还可以辅助评估病情变化。
利益冲突无
猜你喜欢
体育科技文献通报(2022年3期)2022-05-23
运动精品(2022年1期)2022-04-29
医学综述(2021年16期)2021-12-01
自我保健(2020年11期)2021-01-13
中国体育教练员(2018年2期)2018-07-23
运动(2018年14期)2018-07-16
体育科学(2018年3期)2018-04-20
教育教学论坛(2018年1期)2018-01-18
中外医学研究(2017年15期)2017-06-29
求知导刊(2017年6期)2017-04-15
