
FUS基因变异型家族性肌萎缩侧索硬化症一例
2021-09-22 06:28:06李想张慧媛张俭何志义
中国现代神经疾病杂志 2021年6期
李想 张慧媛 张俭 何志义
患者 女性,25岁。主因双下肢无力伴言语不清、吞咽困难9个月,双上肢无力2个月,于2019年3月29日入院。患者9个月前无明显诱因出现右下肢无力,7个月前出现左下肢无力,此后症状进行性加重并伴随言语不清、吞咽困难等症状,偶有饮水呛咳,未予治疗;2个月前出现双上肢无力,为求诊断与治疗,遂至我院就诊,门诊以肢体无力收入院。自患病以来症状平稳,无发热,无头痛、头晕,无意识障碍,无抽搐发作,无恶心、呕吐,无视物不清、视物成双及耳鸣等症状与体征。精神状态尚可,大小便正常,近期体重无异常变化,既往身体健康。其父2006年末(42岁)曾因吞咽困难就医,外院体格检查显示舌肌萎缩,四肢肌力尚可,诊断为运动神经元病,未行基因检测,后因肌力减退、吞咽困难无法进食,2008年末(44岁)死于极度营养不良。患者及其母均否认其他家族成员有类似症状或疾病。
诊断与治疗过程 体格检查:神志清楚,查体合作,构音障碍,语调、语态无明显改变。双侧瞳孔等大、等圆,直径约3 mm,对光反应灵敏,双侧眼球运动正常,无眼震;双侧视力、听力未见明显异常。双侧额纹及鼻唇沟对称,软腭及悬雍垂居中,咽反射正常,伸舌居中,舌肌纤颤,软腭运动减弱。颈部柔软。左侧股四头肌可见肌束震颤。四肢近端肌萎缩,双侧鱼际肌萎缩。左上肢近端肌力3级、远端4级,左下肢近端肌力3级、远端2级,右上肢近端肌力3级、远端4级,右下肢近端肌力3级、远端2级;四肢肌张力正常。双侧肱二头肌、肱三头肌腱反射减弱,双侧跟-膝-腱反射减弱。双侧Babinski征阴性。脑干反射检查咬肌下颌反射阳性,双侧掌颏反射呈阳性。实验室检查:血清钾3.44 mmol/L(3.50~5.30 mmol/L),同型半胱氨酸21.43 mmol/L(4.44~13.56 mmol/L),叶 酸 水 平 为6.72 mmol/L(8.83~60.80 mmol/L),余血清学检查无明显异常。脑脊液检查:压力、常规、生化均未见明显异常。影像学检查:颈椎MRI可见C4-7椎间盘轻度膨出,腰椎、骶尾椎CT平扫未见明显异常。肌电图检查:左侧胸锁乳突肌、右侧肱二头肌、右侧拇短展肌、右侧胫骨前肌呈神经源性损害;双侧腓总神经运动神经传导速度(MNCV)正常,远端潜伏期延长,诱发电位波幅降低;右侧尺神经重复频率电刺激未见明显低频递减及高频递增现象(图1)。基因检测:征得患者及其母亲同意,于入院后第7天晨起采集其外周静脉血各5 ml,采用第二代测序技术(NGS)行基因检测(沈阳金域医学检验所有限公司)。结果显示:患者存在FUS基因外显子15c.1553_1554delinsTT(p.Arg518Ile)杂合突变(图2a),患者母亲FUS基因外显子相应区域无变异(图2b)。分别检索人类基因突变数据库(HGMD;http://www.hgmd.cf.ac.uk/ac/index.php)、千 人 基 因 组 数 据 库(https://www.internationalgenome.org/)和美国国家生物技术信息中心(NCBI)dbSNP147数据库(https://www.ncbi.nlm.nih.gov/snp/),均未收录该变异类型,属于新发变异,为国内外首次报道。生物信息学软件(polymorphism phenotype、Soring intolerant from toleran等)综合分析显示,该变异类型在美国医学遗传学和基因组学会(ACMG)的评级为可能的致病性变异,结合患者临床表现和各项辅助检查结果,最终明确诊断为肌萎缩侧索硬化症(ALS)。治疗原则以药物治疗为主,予以维生素B610 mg/d、甲钴胺0.50 mg/次(3次/d)、叶酸5 mg/次(3次/d)口服,同时辅以口服氯化钾0.50 g/次(3次/d),连续治疗8天症状无明显改善或加重,遂出院。出院后继续口服甲钴胺0.50 mg/次(3次/d)2周,嘱定期复查,出院6个月后电话随访,自诉饮水呛咳、吞咽困难症状略有加重,肢体无力症状无明显变化。

图1 肌电图检查显示,右侧胫骨前肌于静息状态下活跃性失神经电位和大力收缩时运动单位电位均减少Figure 1 Results of EMG of right tibialis anterior muscle showed active denervation potential in resting state and decreased motor unit potential from forced muscle contraction.
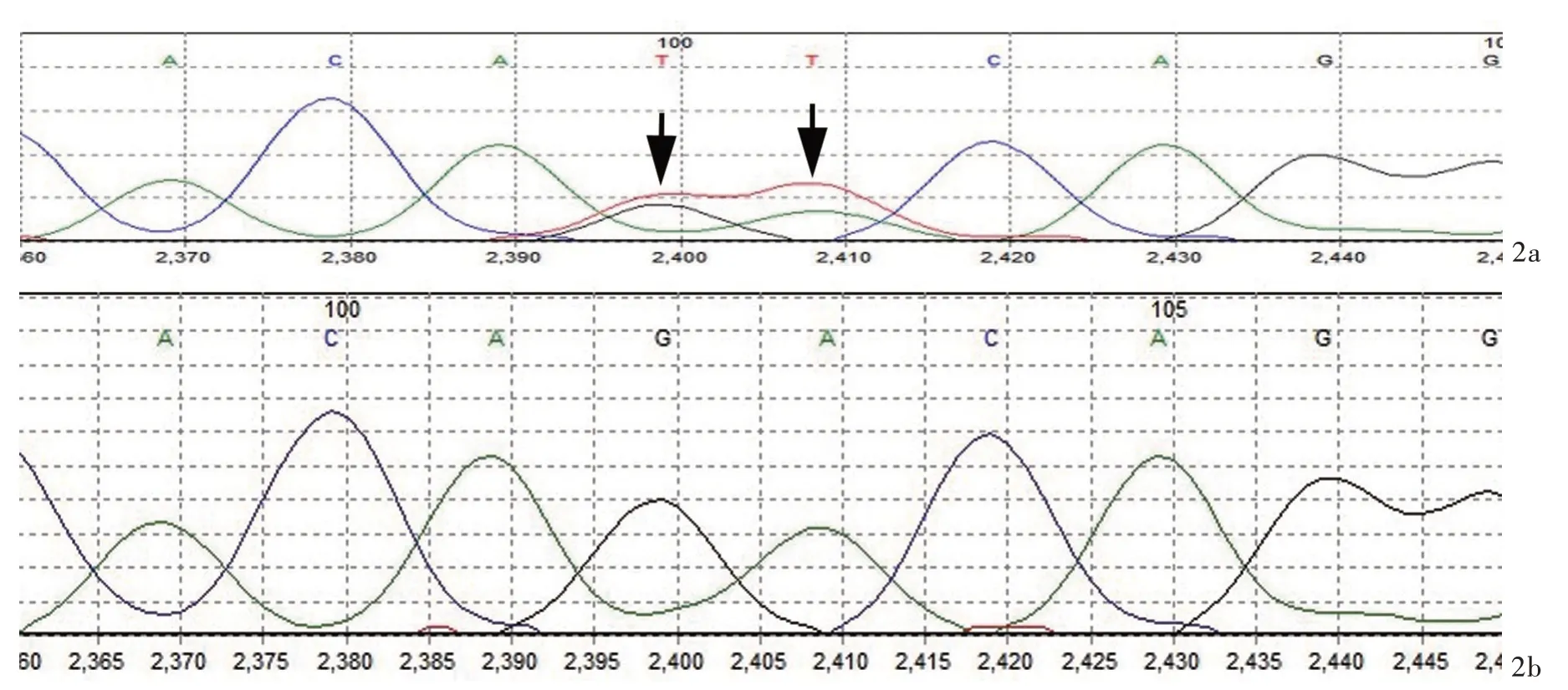
图2 患者及其母基因检测结果 2a 患者存在FUS基因外显子15 c.1553_1554delinsTT(p.Arg518Ile)杂合突变(箭头所示) 2b 患者之母FUS基因外显子相应区域无变异Figure 2 Gene sequencing findings of the patient and her mother There was a missense heterozygous c.1553_1554delinsTT(p.Arg518Ile)mutation in the exon 15 of FUS gene(arrows indicate,Panel 2a).There was no mutation in the corresponding region of the FUS gene exon in the patient's mother(Panel 2b).
讨 论
肌萎缩侧索硬化症是以脑和脊髓运动神经元变性为特征的进行性致死性疾病,是运动神经元病中最为常见的类型,以进行性加重的骨骼肌无力、肌萎缩、肌束颤动、延髓麻痹和锥体束征为主要临床表现,生存期一般为3~5年[1]。本文患者以双下肢无力隐匿发病,呈进行性加重,同时伴有四肢近端肌萎缩、软腭运动减弱、舌肌纤颤、左侧股四头肌肌束震颤、饮水呛咳及吞咽困难,脑干病理反射呈阳性,肌电图提示呈典型的神经源性损害,临床特征较为全面且典型,符合肌萎缩侧索硬化症的临床特征。
约有10%的肌萎缩侧索硬化症患者其一级或二级亲属中存在家族史,这种类型大多归于家族性肌萎缩侧索硬化症(FALS),余则为散发性肌萎缩侧索硬化症(sALS)[2-4]。多数家族性肌萎缩侧索硬化症为常染色体显性遗传,也可有常染色体隐性遗传和X连锁显性遗传[3]。家族性肌萎缩侧索硬化症患者的家系常由1例先证者和1~2例患病一级或二级亲属组成,超过50%的家系仅包括2~3例肌萎缩侧索硬化症病例,多例患病个体家系仅占少数[2,5]。本文报告1例肌萎缩侧索硬化症患者,虽然患者父亲未行基因检测,但根据其临床表现及外院诊断,仍提示该例患者存在阳性家族史,但因患者之子尚年幼(3岁),患者及家属拒绝对其子进行基因检测,故不能证实是否遗传至下一代。
1993年,Rosen等[6]发现,家族性肌萎缩侧索硬化症患者存在SOD1基因相关变异,随着对肌萎缩侧索硬化症遗传学研究的不断深入,目前已鉴定逾50种潜在的致病或疾病修饰基因,其中C9ORF72、SOD1、TARDBP、FUS和ANG变异占肌萎缩侧索硬化症患者变异基因的50%以上[2-3,7-8],其他基因则相对罕见。一项包含欧洲和亚洲人群的系统回顾分析表明,在家族性肌萎缩侧索硬化症和散发性肌萎缩侧索硬化症患者中,主要肌萎缩侧索硬化症相关基因的总变异频率分别为47.7%和5.2%,但欧洲和亚洲患者的主要肌萎缩侧索硬化症基因变异频率存在显著差异;欧洲人群以C9ORF72重复扩增最为常见,FUS基因变异位列第四,而在亚洲人群中,以SOD1变异最为常见,其次是FUS变异[9]。其他相关研究也得出相似结论[10-11],提示中国大陆地区FUS变异频率增高,是区别于白种人的遗传特征之一。因此,本文患者作为存在FUS基因变异的家族性肌萎缩侧索硬化症患者,虽在家族性肌萎缩侧索硬化症总体基因变异类型分布中属于相对罕见类型,但符合中国大陆地区的遗传变异特征。
FUS基因变异定位于染色体16p11.2,通常以常染色体显性方式遗传,约占家族性肌萎缩侧索硬化症的4%,在散发性肌萎缩侧索硬化症患者中则较为少见[12-16]。研究表明,超过60%的FUS基因变异型肌萎缩侧索硬化症患者在45岁以前发病[17]。近年有研究发现,FUS基因变异与青少年肌萎缩侧索硬化症(JALS),尤其是散发性青少年肌萎缩侧索硬化症密切相关[18],同时该项研究指出,由FUS基因变异导致的散发性青少年肌萎缩侧索硬化症患者,与其他基因变异类型相比,其病程进展更快、生存期更短。我国一项针对12例青少年肌萎缩侧索硬化症患者的研究显示,有30%(4例)的患者存在FUS基因变异,表明FUS基因变异可能是肌萎缩侧索硬化症早发遗传的关键影响因素[19]。2020年,Chen等[20]对我国南部地区268例肌萎缩侧索硬化症患者基因型与表型进行研究,其中132例(49.25%)存在肌萎缩侧索硬化症相关基因变异,FUS基因变异者4例,占总变异数的3.03%,此4例患者男女比例1∶1,平均发病年龄(33.5±12.4)岁,在所有基因变异类型中发病最早,且与总体平均发病年龄[(52.1±10.4)岁]相比有显著差异(P=0.001),提示FUS基因变异型肌萎缩侧索硬化症患者更倾向于早发;该项研究所有FUS基因变异患者均从肢端发病(3例上肢、1例下肢),而无球部发病。本文患者虽不属于青少年肌萎缩侧索硬化症,但其发病年龄相对较早,以双下肢无力发病,这些特征与上述研究得出的“早发性”和“肢端发病”结论可以相互印证。
FUS基因编码的肉瘤融合/脂肪肉瘤转运蛋白(FUS/TLS)是一种在神经元中普遍表达的蛋白质,主要位于胞核内[12],也可存在于神经元树突中[21];属于不均一核糖核蛋白(hn-RNP)复合体的组成成分之一,由526个氨基酸组成,15个外显子编码,其相对分子质量为75×103[22],其可通过与运动蛋白驱动蛋白[23]和肌球蛋白-Va[24]相结合参与DNA和RNA的代谢过程[12,25]。其N-端具有转录激活域,在染色体易位后可产生导致尤文肉瘤和急性髓性白血病的融合蛋白[26];C-端含有RNA识别基序(RRM)、精氨酸/甘氨酸/甘氨酸重复序列和参与RNA加工的锌指结构域,以及一个高度保守的极限C-端区[12,22]。FUS/TLS蛋白变异点即位于此C-端高度保守的RNA结合结构域中[12],这种缺陷蛋白大多在神经元的细胞质内聚集。研究发现,染色体16p连锁的FUS基因错义突变与家族性肌萎缩侧索硬化症相关[12,27]。目前发现的FUS基因变异形式已经超过50种,主要集中于外显子6、14和15,其中大部分为错义突变,少数是移码或无义突变[28-29]。本文患者基因变异位点即定位于染色体16p11.2,为错义突变,在外显子15中检测到单个碱基对变化,这种变化导致在518位置处的精氨酸被异亮氨酸所取代(p.Arg518Ile)。目前比较明确的错义突变有p.Arg514Ser、p.Pro525Leu[30],以 及p.Arg521Cys、p.Arg521His[31]等。既往报道显示,外显子15为变异热区,且此前已有研究者检测到与本文患者位置相同的第518位氨基酸变异[32],但Arg518Ile变异尚未见诸报道。合并FUS基因Arg518Ile变异的肌萎缩侧索硬化症患者在表型上有哪些更细微的特征,尚待进一步探究。此外,FUS/TLS蛋白亦是亨廷顿舞蹈病[33]、额颞叶痴呆[21]等其他神经退行性疾病的主要聚集作用蛋白。FUS基因的异常表达亦与黏液样脂肪肉瘤[34]、前列腺癌[35]、非小细胞肺癌[36]等肿瘤的发病机制密切相关,提示FUS基因变异在肌萎缩侧索硬化症、其他神经退行性疾病以及肿瘤等均具有可能的致病作用,并为进一步探索FUS基因的表达作用提供了广阔思路。
本文报告1例FUS基因变异型家族性肌萎缩侧索硬化症患者,属于肌萎缩侧索硬化症之少见类型。FUS基因变异型在家族性肌萎缩侧索硬化症人群中所占比例也相对较小,但由于该基因在中国大陆突变频率增高,并成为区别于白种人的遗传特征之一,故报告该病例有助于提高医学工作者对FUS基因变异型家族性肌萎缩侧索硬化症的认识,更精准地进行临床诊治。
利益冲突无
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29 01:57:52
中国生殖健康(2020年4期)2021-01-18 02:58:10
中国临床医学影像杂志(2019年5期)2019-08-27 02:47:44
中国生殖健康(2018年4期)2018-11-06 07:12:16
高中生·天天向上(2017年4期)2017-06-09 02:37:17
中西医结合心脑血管病杂志(2016年20期)2016-03-01 04:20:48
西安交通大学学报(医学版)(2015年2期)2015-02-28 17:59:20
湖北农业科学(2014年11期)2014-09-10 18:06:07
河北医科大学学报(2010年10期)2010-03-25 10:14:57
河北医科大学学报(2010年10期)2010-03-25 10:14:55
