
HPLC法测定大黄碳酸钠片中非法成分土大黄苷的含量及UPLC-MS/MS确证
2021-09-13 02:34黄春晖胡军廖乃英
右江医学 2021年7期
黄春晖 胡军 廖乃英

【摘要】 目的 建立高效液相色譜法(HPLC)测定大黄碳酸钠片的组方药材大黄伪品中指标性成分土大黄苷的含量,并建立超高效液相色谱-串联质谱法(UPLC-MS/MS)确证方法。
方法 HPLC法采用色谱柱为CAPCELL PAK MGⅡ(4.6 mm×250 mm,5 μm),流动相为乙腈-水(20∶80),检测波长为325 nm,流速为1.0 mL·min-1,进样量为10 μL。UPLC-MS/MS确证方法采用色谱柱为Agilent Zorbax Eclipse Plus C18(2.1 mm×100 mm,1.8 μm),流动相为0.1%甲酸水溶液-乙腈(80∶20),流速为0.2 mL·min-1,柱温为40℃,进样量为2 μL;采用电喷雾离子源(ESI)在负离子模式下,利用多反应监测(MRM)模式对土大黄苷进行分析,定量及定性离子对分别为m/z 418.9→257.1和m/z 418.9→160.9。
结果 HPLC法方法学考察结果显示,土大黄苷在0.59~23.56 μg·mL-1质量浓度范围内与峰面积线性关系良好(r=0.9997),检出限为0.06 μg·mL-1,平均加样回收率为100.3%(n=9,RSD=1.5%)。12个批次的大黄碳酸氢片中均未检出土大黄苷成分。UPLC-MS/MS法检测结果显示,土大黄苷在0.024~1.178 μg·mL-1质量浓度范围内与峰面积线性关系良好(r=0.9996),检出限为0.7 ng·mL-1,平均加样回收率为99.4%(n=9,RSD=1.5%)。12个批次的大黄碳酸氢片中均未检出质荷比(m/z)418.9的准分子离子峰和m/z 257.1、160.9的碎片离子峰。
结论 所建立的HPLC法与UPLC-MS/MS法操作简便、快速灵敏,结果准确,可作为大黄碳酸钠片中非法成分土大黄苷的质量控制方法。
【关键词】 高效液相色谱法;超高效液相色谱-串联质谱法;大黄碳酸钠片;非法成份;土大黄苷;含量测定
中图分类号:R282.5 文献标志码:A DOI:10.3969/j.issn.1003-1383.2021.07.002
【Abstract】 Objective To establish high performance liquid chromatography (HPLC) for the determination of rhaponiticin illegally added in rhubarb and sodium bicarbonate tablets, and to establish ultra performance liquid chromatography and tandem mass spectrometry (UPLC-MS/MS) for the identification of rhaponiticin.
Methods In HPLC method, chromatographic column was CAPCELL PAK MGⅡ(4.6 mm × 250 mm, 5 μm), mobile phase was acetonitrile-water (20∶80), detection wavelength was 325 nm, velocity of flow was 1.0 mL·min-1, and injection volume was 10 μL. In UPLC-MS/MS corroboration method, chromatographic column was Agilent Zorbax Eclipse Plus C18 (2.1 mm × 100 mm, 1.8 μm), mobile phase was 0.1% methyl acid aqueous solution-acetonitrile (80∶20), velocity of flow was 0.2 mL·min-1, column temperature was 40℃, injection volume was 2 μL. Electric spray ion source (ESI) negative ion mode and multiple reaction monitoring (MRM) were used to analyze rhaponiticin, and quantitative and qualitative ion pairs were m/z 418.9 → 257.1 and m/z 418.9 → 160.9, respectively.
Results Results of HPLC methodology investigation showed that within mass concentration range of 0.59-23.56 μg·mL-1, rhaponiticin had good relation with peak area (r=0.9997), detection limit was 0.06 μg·mL-1, average recovery rate of adding sample was 100.3% (n = 9, RSD = 1.5%). Results of UPLC-MS/MS detection showed that within mass concentration range of 0.024 -1.178 μg·mL-1, rhaponiticin had good relation with peak area ( r = 0.9996), detection limit was 0.7 ng·mL-1, average recovery rate of adding sample was 99.4% (n = 9, RSD=1.5%). No quasimolecular ions with m/z of 418.9 and fragment ions with m/z 257.1 and 160.9 were detected in 12 batches of rhubarb and sodium bicarbonate tablets.
Conclusion HPLC and UPLC-MS/MS methods are simple, rapid, sensitive and accurate, which can be used for the quality control of rhaponiticin illegally added in rhubarb and sodium bicarbonate tablets.
【Key words】 HPLC; UPLC-MS/MS; rhubarb and sodium bicarbonate tablet; illegal components; rhaponiticin; content determination
大黄碳酸氢钠片属于中西药复方制剂,其主要成分为大黄、碳酸氢钠和薄荷油[1],用于治疗食欲不振、胃酸过多、消化不良和便秘等。大黄作为大黄碳酸氢钠片处方中的君药,其主要作用是引起唾液和胃液的分泌增多,使食欲增加,胃肠轻度充血,加强肠胃吸收功能[2],临床主要用于治疗积滞便秘、血热呕吐、目赤咽肿、热毒疮疡、烧烫伤、血瘀证等[3~5]。大黄药材应不含土大黄苷成分,而伪品大黄中含有土大黄苷[6~7]。大黄碳酸氢钠片现行标准中只有碳酸氢钠的含量测定,没有大黄药材的定性或定量的测定,无法监督检查企业投料大黄的真伪情况。当前市场上大黄有用华北大黄、河套大黄、臧边大黄等伪品大黄掺假或冒充使用的情况[8]。因此,有必要对大黄碳酸氢钠片中的土大黄苷成分进行严格的筛查,增强该产品的安全性。土大黄苷属二苯乙烯苷类成分,目前主要的检测方法有薄层色谱法(TLC)[9~10]、高效液相色谱法(HPLC)[11]和液相-液质联用法(LC-MS)[12~13]。由于TLC法属于定性鉴别,容易出现假阳性现象。而HPLC法能根据保留时间和DAD光谱进行定性和定量测定,但是结果易受辅料干扰,灵敏度较LC-MS法差。LC-MS法具有快速、准确、简便、全面、灵敏度高、选择性好等优点,可以通过一级质谱和二级质谱对测定结果进行确证。为了有效控制大黄碳酸氢钠片产品质量,保证用药安全,本文建立了HPLC法和超高效液相色谱-串联质谱法(UPLC-MS/MS)对大黄碳酸钠片中非法成分土大黄苷进行筛查,为该制剂的质量控制提供参考。
1 材料与方法
1.1 仪器及材料
Waters2695-2998高效液相色谱仪,Agilent1290超高压液相色谱仪(美国安捷伦公司),6460 QQQ三重四级杆质谱仪(美国安捷伦公司),CAPCELL PAK MGⅡ(4.6 mm×250 mm,5 μm)色谱柱,Agilent Zorbax Eclipse Plus C18(2.1 mm×100 mm,1.8 μm)色谱柱,XD205DU分析天平(梅特勒公司)。
1.2 药品
土大黄苷对照品(中国食品药品检定研究院,批号110794-201708,供TLC和HPLC检查用),大黄碳酸氢钠片(①广西十万山制药有限公司,批号181202、181003、180305;②广西金页制药有限公司,批号190608、190808;③广西北部湾制药股份有限公司,批号180203、180201、180202;④四川德元药业集团有限公司,批号180903、180904;⑤湖南福来药业有限公司,批号190403、190404)购自南宁市区药店。乙酸铵、乙腈(均购自德国Merck公司,含量均为99.9%),水为超纯水。
1.3 HPLC色谱条件
色谱柱为CAPCELL PAK MGⅡ(4.6 mm×250 mm,5 μm),流动相为乙腈-水(20∶80),检测波长为325 nm,流速为1.0 mL·min-1,进样量为10 μL。
1.4 UPLC-MS/MS色谱质谱条件
色谱柱为Agilent Zorbax Eclipse Plus C18(2.1 mm×100 mm,1.8 μm);流动相为0.1%乙酸水溶液-乙腈(80∶20),流速为0.2 mL b min-1,柱温40℃,进样量为2 μL。采用电喷雾离子源(ESI)在负离子模式下,利用多反应监测(MRM)模式。离子源温度350℃;雾化器压力40 Psi;干燥气流速:12 L·min-1,干燥气温度250℃,碎裂电压为100 V,碰撞能量为25 V。定量及定性離子对分别为m/z 418.9→257.1和m/z 418.9→160.9。
1.5 溶液的制备
(1)对照品溶液的制备:取土大黄苷对照品11.78 mg置于100 mL容量瓶中,加流动相溶解并稀释至刻度。作为对照品储备液。精密量取对照品储备液1 mL置50 mL量瓶中,加流动相稀释至刻度,摇匀,即得土大黄苷浓度为2.356 μg·mL-1。(2)供试品溶液的制备:取大黄碳酸氢钠片样品20片,研细后精密称取细粉适量,加流动相溶解稀释制成含大黄3 mg·mL-1的溶液,摇匀,滤过,即得。(3)阴性样品溶液的制备:按处方制备不含大黄的阴性样品,混合均匀后按供试品溶液制备即得。
2 结 果
2.1 HPLC测定结果
2.1.1 专属性考察
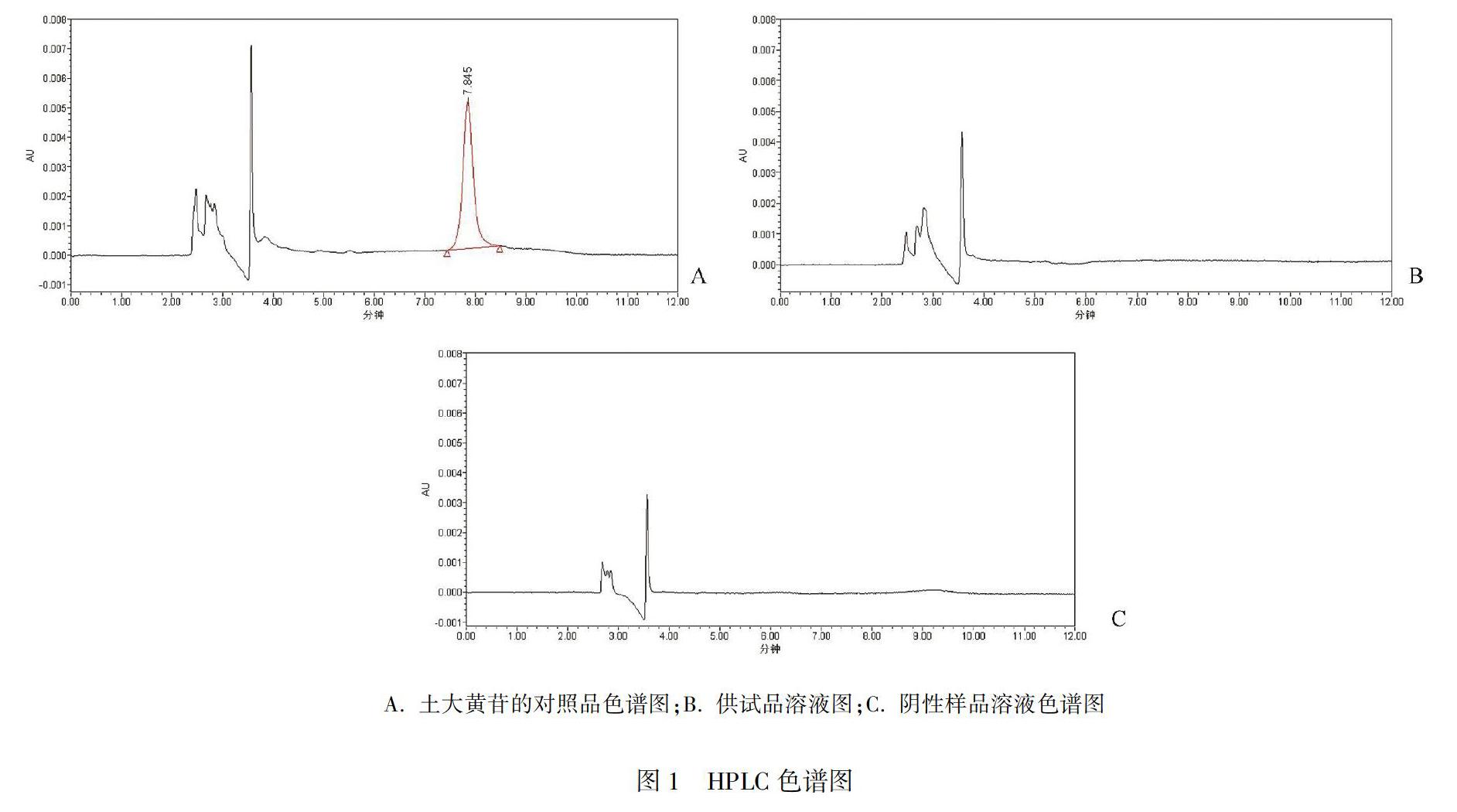
取“1.5”项下的对照品溶液、供试品溶液、阴性样品溶液各10 μL,按“1.3”项下的色谱条件进行测定,记录色谱图,结果见图1。对照品溶液中土大黄苷的保留时间为7.8 min,阴性样品溶液不影响结果测定,12个批次的大黄碳酸氢片的供试品溶液中均未检出土大黄苷成分。
2.1.2 线性关系考察
分别精密移取“1.5”项下的对照品储备液0.25、0.5、1.0、2.0、5.0、10 mL置50 mL容量瓶中,用流动相稀释刻度,即得系列标准曲线溶液。按“1.3”项下色谱条件进样测定,以土大黄苷的浓度(X,μg·mL-1)作为横坐标,以峰面积(Y)作为纵坐标。结果表明土大黄苷在0.59~23.56 μg·mL-1范围内线性关系良好,回归方程为Y=32 164.42X-5871.72(r=0.9997,n=6)。
2.1.3 检测限与定量限
取“2.1.2”项下曲线溶液1,进样测定,记录信噪比。结果土大黄苷的检出限为0.06 μg·mL-1(S/N=3),定量限为0.19 μg·mL-1(S/N=10)。
2.1.4 进样精密度和稳定性试验
取“1.5”项下制备的对照品溶液连续进样6次,记录峰面积,结果土大黄苷峰面积的RSD为1.1%(n=6)。取“1.5”项下制备的对照品溶液,分别于室温放置0、2、4、8、12、18、24 h时进样测定,记录峰面积,结果土大黄苷峰面积的RSD为1.3%(n=7),表明土大黄苷在室温下放置24 h内稳定性良好。
2.1.5 加样回收率试验
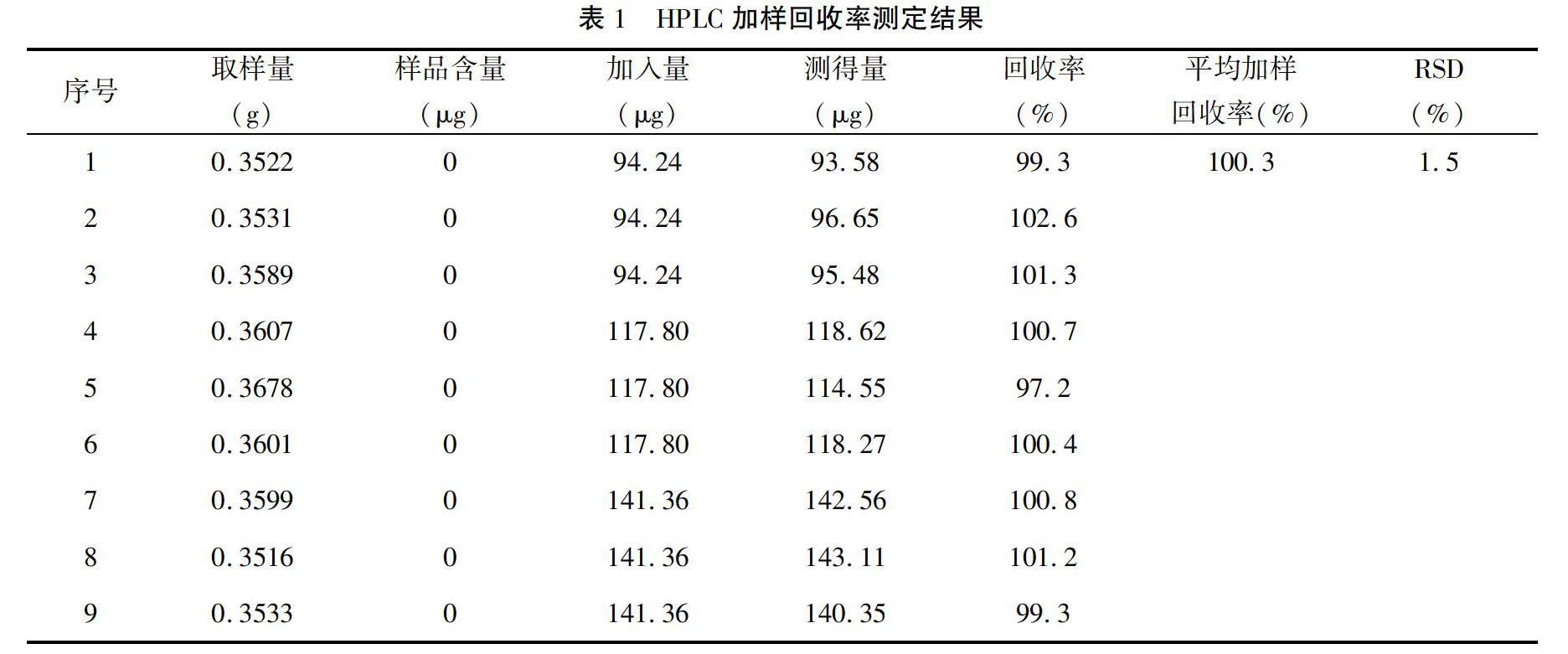
取大黄碳酸氢钠片20片,精密称定,研细,精密称取细粉约1片的重量(约相当于大黄150 mg),置50 mL量瓶中,加流动相使其溶解,再分别精密加入“1.5”项下制备的对照品溶液储备液0.8、1.0、1.2 mL,每个水平浓度各3份,再用流动相稀释至刻度,按“1.3”项下色谱条件进样测定,记录峰面积并计算加样回收率,结果见表1。平均回收率为100.3%,RSD为1.5%,符合方法学要求。
2.1.6 耐用性试验
进行预实验比较不同厂家色谱柱的分离效果。色谱柱A:CAPCELL PAK MGⅡ(4.6 mm×250 mm,5 μm);色谱柱B:Waters HSS-C18(4.6 mm×250 mm,5 μm);色谱柱C:Agilent SB-C18(4.6 mm×250 mm,5 μm),结果发现色谱柱A得到的土大黄苷峰型最好,与相邻峰分离效果最好,理论塔板数最高,因此选择色谱柱A作为分析色谱柱。
2.1.7 样品含量的测定
取5个不同生产厂家共12批次的样品,按“1.5”项下方法制备供试品溶液,再按上述色谱条件进样测定,记录峰面积并计算样品中土大黄苷的含量。结果12批次的样品中均未检出土大黄苷,说明生产企业未存在违规投料现象。
2.2 UPLC-MS/MS对检测结果的确证
2.2.1 溶液的制备
(1)对照品溶液的制备:取“1.5”项下的对照品储备液稀释制成含土大黄苷浓度为2.356 μg·mL-1的对照品使用液。(2)供试品溶液的制备:取“1.5”项下供试品溶液,稀释10倍后用0.22 μm微孔滤膜滤过,即得。(3)阴性样品溶液的制备:取“1.5”项下的阴性样品溶液,稀释10倍后用0.22 μm微孔滤膜滤过,即得。
2.2.2 系统适用性试验
对照品溶液在负离子模式下通过MSMS二级质谱扫描发现,土大黄苷得到3个碎片离子(见图2A),其中m/z 257.1和m/z 160.9离子丰度比最大,所以选择m/z 257.1和m/z 160.9作为定量离子。将“2.2.1”项下的对照品溶液、供试品溶液和阴性样品溶液,按“1.4”项下的质谱条件进样测定,得到质谱的TIC总离子流图(见图2B、C、D)。结果显示,在该色谱条件下,土大黄苷在1.8 min左右出峰,且峰型良好。样品未检出土大黄苷的定量及定性离子对,阴性样品溶液对测定无干扰。
2.2.3 线性关系考察
精密吸取“2.2.1”项下制备的对照品使用液0.1、0.2、0.5、1.0、2.0、5.0 mL,置于不同10 mL量瓶中,加20%乙腈溶液稀释至刻度,摇匀,制成标准曲线溶液。按“1.4”项下的色谱质谱条件进样测定,记录峰面积。以对照品的质量浓度(x,μg·mL-1)为横坐标,峰面积(y)为纵坐标进行线性回归。结果土大黄苷在0.024~1.178 μg·mL-1质量浓度范围内与峰面积呈良好的线性关系,回归方程为:y=109 243.89x+895.91(r=0.9996)。
2.2.4 定量限(LOQ)与检测限(LOD)考察
取“2.2.1”项下制备的对照品使用液适量,逐级稀释,分别进样,以信噪比为10左右计算定量限,以信噪比为3左右计算检测限,结果土大黄苷的检出限为0.7 ng·mL-1,定量限为2.3 ng·mL-1。
2.2.5 加样回收率试验
取大黄碳酸氢钠片10片,精密称定,研细,精密称取细粉约1片的重量(约相当于大黄150 mg),置50 mL量瓶中,加20%乙腈溶液使溶解并稀释至刻度,摇匀,滤过,精密量取续滤液1 mL置10 mL量瓶中,再分别精密加入“2.2.1”项下制備的对照品使用液0.8、1.0、1.2 mL,每个水平浓度各3份,再用20%乙腈溶液稀释至刻度,按“1.4”项下色谱质谱条件进样测定,记录峰面积并计算加样回收率,结果见表2。平均回收率为99.4%,RSD为1.5%,符合方法学要求。
2.2.6 样品含量的测定
取大黄碳酸氢钠片供试品溶液与土大黄苷对照品溶液,再按上述色谱条件进样测定,记录一级质谱图与二级质谱图。结果显示,在土大黄苷对照品稀释液中检测到其准分子离子[M-H]-峰质荷比(m/z)为418.9;对m/z 418.9峰进行二级子离子扫描,产生的主要碎片离子m/z分别为257.1、160.9。供试品溶液经检测均未产生与土大黄苷对照品一致的准分子离子,这进一步证实了12批次的样品中不含土大黄苷成分。
3 讨 论
3.1 质谱条件的优化
(1)扫描模式的选择:土大黄苷的分子量为420.41,在正离子模式下进行全扫描可以得到[M+H]+母离子为421.3,在负离子模式下进行全扫描可以得到[M-H]-母离子为418.9。比较两种模式下两个母离子的响应值,结果负离子模式下离子的响应值较大,因此选择负离子模式。(2)碎裂电压与定碰撞能量的选择:在负离子模式下通过选择离子扫描(sim),分别考察在碎裂电压分别为130、120、110、100、90、80、70 V时得到的母离子的响应值,结果发现在100V时离子的响应值最大,因此选择碎裂电压为100 V。在碎裂电压为100 V的条件下,进行子离子扫描,分别考察碰撞能为0、5、10、15、20、25、30 V时得到的碎片离子的响应值,结果发现在碰撞能量为25V时,两个子离子m/z 257.1和m/z160.9响应值最大。
3.2 两种方法比较
HPLC法与UPLC-MS/MS法均可对大黄碳酸钠片中非法成分土大黄苷的含量进行测定,但是HPLC法仅涉及定量测定,无法对其质量数进行确证,因此容易出现假阳性现象。遇到可能存在非法添加的样品时,需要辅助UPLC-MS/MS法,通过一级质谱和二级质谱扫描对其质量数进行确证。同时,UPLC-MS/MS技术具有灵敏度高、检出限低、样品用量少的优点,其质谱检测器相比HPLC的紫外检测器大幅度提高了灵敏度和选择性,对于含量比较低的土大黄苷定量限提高了83倍,也避免了大黄碳酸钠片中其他成分的紫外干扰,大大增加了方法的适用性和灵活性。
本研究建立了HPLC法测定大黄碳酸钠片中非法成分土大黄苷的含量,并利用UPLC-MS/MS对其进行结构确证,两种方法快速简便、全面准确、灵敏度高、选择性好,能准确判定供试品中是否含有土大黄苷,可为大黄碳酸氢钠片监督管理工作提供强有力的技术保障。
参 考 文 献
[1] 金鹏,吴旭.大黄碳酸氢钠片评价性抽验结果及质量评价[J].中国药物评价,2017,34(4):300-304.
[2] 袁爱.大黄碳酸氢钠和复方氢氧化铝在消化系统中的作用比较[J].臨床医药文献电子杂志,2017,4(17):3365-3366.
[3] 郭兴蕾,徐海星,许沛虎,等.不同产地大黄红外指纹图谱及相似度分析[J].中国药师,2018,21(7):1174-1176.
[4] 吕晋,王黛莹.利用近红外光谱法建立大黄药材一致性检验模型[J].中国药师,2018,21(6):1128-1130.
[5] 黄良永,郑江萍,梁俊,等.大黄配方颗粒蒽醌成分的HPLC指纹图谱研究[J].中国药师,2014,17(8):1305-1308.
[6] 陈峰.不同种大黄中土大黄苷检查方法的研究[J].中国民族民间医药,2018,27(5):14-16.
[7] 冯有龙,余伯阳.HPLC法鉴别大黄及部分含大黄中成药的真伪[J].中国药品标准,2009,10(4):296-298.
[8] 任伟光,王琦,黄林芳.大黄类药材的质量评价进展[J].中南药学,2014,12(4):354-359.
[9] 严华,魏锋,肖新月,等.同属不同种大黄及含大黄制剂中土大黄苷检查方法的研究[J].药物分析杂志,2010,30(9):1615-1620.
[10] 王银红,万胜利,王艳,等.TLC、HPLC、UPLC-MS法联合定性检查厚朴排气合剂中的土大黄苷[J].中南药学,2013,11(10):764-766.
[11] 蒋永海,朱辉.妇炎消胶囊中非法成分土大黄苷的检测[J].中国现代应用药学,2011,28(10):959-961.
[12] 孙爱萍,张西如,谷菲菲.LC-MS/MS方法测定中成药麻仁丸中添加伪品大黄的研究[J].中成药,2011,33(2):357-359.
[13] 陈学艳,张敏娟,魏文芝.复方龙胆碳酸氢钠片中非法成分土大黄苷定性筛查与定量测定方法的建立[J].中国药房,2019,30(14):1919-1924.
(收稿日期:2021-03-22 修回日期:2021-05-07)
(编辑:潘明志)
猜你喜欢
分析化学(2017年1期)2017-02-06
中国纤检(2016年12期)2017-01-20
中国民族民间医药·上半月(2016年11期)2016-12-26
中国民族民间医药·上半月(2016年11期)2016-12-26
考试周刊(2016年95期)2016-12-21
上海医药(2016年1期)2016-02-22
安徽农学通报(2015年10期)2015-06-15
分析化学(2014年3期)2014-12-16
现代仪器与医疗(2014年1期)2014-03-28
