
基于BSA和SLAF-Seq技术对大豆主茎节数QTL精细定位
2021-09-13 07:27:52杨玉花白志元卫保国张瑞军
核农学报 2021年9期
杨玉花 白志元 卫保国 雷 阳 张瑞军
(1山西农业大学农业基因资源研究中心/农业部黄土高原作物基因资源与种质创制重点实验室/杂粮种质资源发掘与遗传改良山西省重点实验室,山西 太原 030031;2山西农业大学园艺学院,山西 太原 030031)
大豆[Glycine max(L.)Merr.]是世界上重要的农作物之一,也是食用油、植物蛋白和饲料的主要来源,由于目前大豆的供需极度不平衡,因此高产优质是大豆育种的最重要目标[1]。大豆主茎节数是从子叶节以上至主茎顶端的有效节数,是一个相对稳定的数量性状,且遗传力较高[2]。该性状不仅是影响大豆株型的主要性状,同时也是影响大豆产量的重要农艺性状之一。因此,应用高通量测序技术快速精准定位大豆主茎节数相关位点,开发与大豆主茎节数位点紧密连锁的且易于操作和低成本的分子标记,从而加速大豆主茎节数分子标记辅助育种,促进大豆高产优质育种产业的快速发展。传统遗传学方面得到一定的研究进展,如王芳[3]研究发现大豆主茎节数与单株荚数和每荚粒数等产量农艺性状相关性较高,暗示通过更直观的主茎节数也可以洞察植株产量等性状。李玉清[4]研究发现大豆主茎节数性状具有一定的母体效应,可以早代选择。同时前人在大豆主茎节数数量性状(quantitative trait locus,QTL)分子遗传定位方面做了很多研究,目前有37 个与大豆主茎节数相关的QTL定位结果(https:/ /soybase.org/)[5-12]。如Zhang 等[7]以Kefeng 1 号和Nannong1138-2 为亲本杂交构建含有184 个株系的重组自交系(recombinant inbred lines-RIL) 群体,利用限制性内切酶片段长度多态性(restriction fragment length polymorphism,RFLP)、简单重复序列标记(simple sequence repeat,SSR)和表达序列标签(expressed sequence tag,EST)标记构建遗传图谱,共检测到10 个与大豆主茎节数相关的QTL。Chen等[8]以美国大豆品种Charleston×东农594 的F2:10代154 个株系的重组自交系为材料,检测到7 个主茎节数QTL。Liu 等[9]利用Jinpumkong 2×SS2-2 (J×S)和Iksannamulkong×SS2-2 (Ⅰ×S)两个重组自交系群体构建遗传连锁图,共检测到2 个主茎节数QTL。由前人研究报道可知,大豆主茎节数的QTL 定位研究较少,并且对大豆主茎节数分子方面的研究较浅薄。由于前人定位所采用的标记大多数是SSR 标记(可开发的大豆SSR 标记较少),所以定位的区间都比较大。随着大豆全基因组测序信息的公布,利用重测序技术可以更精确检测与主茎节数关联位点和基因,重测序紧密连锁的分子标记是单核苷酸多态性(single nucleotide polymorphism,SNP),但是SNP 标记不能直接运用,必须基于SNP 多态转化成酶切扩增多态性标记(cleaved amplified polymorphic sequences,CAPS)或者衍生 CAPS 标记( derived cleaved amplified polymorphic sequences,dCAPS)才能应用。但由于标记的转化过程耗时长,且转化成本高,因此利用双亲间的插入缺失核酸位点设计InDel 标记作为与大豆主茎节数紧密连锁的分子标记具有可行性,并且InDel 标记与SSR 标记同样具有变异稳定,多态性强,成本低的优点。
本研究以大豆品种中119 和C025 为亲本构建RIL 群体,利用传统分群分析法(bulk segregant analysis,BSA)和全基因组特异性位点扩增片段测序手段(specific-locus amplified fragment sequencing,SLAF-Seq)关联与大豆主茎节数相关位点。同时利用双亲的25×重测序,检测双亲在关联区间的InDel遗传变异。依据关联区间开发的InDel 标记信息对主效区间进行精细定位。本研究结合BSA-SLAF-Seq高通量测序技术和亲本重测序信息,并开发InDel 标记加密关联区间,旨在实现大豆主茎节数位点的精细定位,为大豆主茎节数候选基因的克隆奠定基础,同时加速定向改良大豆主茎节数分子标记辅助育种进程。
1 材料与方法
1.1 试验材料田间试验和性状考察
本研究利用山西省农业科学院农作物品种资源研究所收集的C025(少主茎节数)大豆为母本,中119(多主茎节数)大豆为父本杂交获得F1,自交获得188个单株的F2群体,并记录F2表型。然后通过5 代自交获得102 个单株/系的RIL 分离群体。2017年和2018年在山西省农业科学院东阳基地种植父母本、F1和RIL 群体并考察主茎节数性状。田间试验按照完全随机区组设计,3 次重复,每个小区2 行,株距为50.0 cm,行距为13.5 cm。大豆成熟时,每个小区选择10个代表单株人工统计主茎节数(从主茎节到顶端之间的所有节数),并计算其平均值[13]。
1.2 SLAF 文库构建测序以及SLAF 标签的开发和SNP 标记检测
利用北京百迈客生物科技有限公司自主研发的酶切预测软件对大豆参考基因组(Wm82.a2.v1)进行酶切预测,最终确定本研究采用的限制性内切酶为RsaI+HaeIII,对检测合格的4 个样品(父、母本,少主茎节混池和多主茎节混池)基因组DNA 分别进行酶切。然后通过PCR 扩增、纯化、混样、切胶选取目的片段等,最后利用Illumina HiSeqTM2500 平台完成测序。
特异长度扩增片段测序(specific length amplified fragment sequencing,SLAF-Seq)测序数据分析参照Sun等[14]操作程序处理。基于序列相似度对所有具有清晰索引信息的SLAF 双端读取进行聚类,并在BLAST 中采用一对一的方法进行检测,结果显示平均比对效率均在91.47%以上,说明本研究各个样品测序数据正常。通过3′端加A 处理、连接Dual-index[15]测序接头对每个样品的原始数据进行处理。通过评估序列质量和数据量对原始数据进行过滤。然后将clean reads 与参考基因组数据进行比对,并开发SLAF 标签和SNP 标记[16]。通过相关分析确定与大豆主茎节数密切相关的SNP,并根据相关阈值确定候选区间。最后,进行基因功能注释和生物途径富集分析,以确定候选区间的基因。
1.3 ED 值和SNP-index 关联分析
依据RIL 群体30 株极端多主茎节数(用RR 表示)和30 株极端少主茎节数(用Rr 表示)混池表型,以及SLAF 测序后的基因型,利用欧式距离(euclidean distance,ED)算法,寻找两个混池间存在显著差异标记,从而评估与主茎节数性状的关联区间。理论上,除了BSA 测序建立的两个混合基因库中与目标性状相关的基因座存在差异外,其他基因座的差异趋于一致,因此,非目标相关基因座的ED 值等于0。ED 值估算如下:ED=
式中,ARR、TRR、CRR、GRR分别代表RR 混池中各个碱基的频率,而ARr、TRr、CRr、GRr分别代表在Rr 混池中各个碱基的频率。理论上ED 值越高,越接近目标位点[17]。
在进行分析时,利用两个混池间基因型存在差异的SNP 位点,统计各个碱基在不同混池中的深度,并计算每个位点ED 值[18]。本研究将ED 的2 次方作为关联值以达到消除背景噪音的功能,然后采用SNPNUM 方法对ED 值进行拟合,并根据关联阈值选择阈值以上的区间作为与主茎节数基因相关的区间。
同时采用另一种ΔSNP-index 算法评估与主茎节数性状分离相关的位点。SNP-index 统计公式如下:
Maa 和Paa 分别是表示父本和母本的测序深度,Mab 和Pab 分别表示混池性状与母本一致的和混池性状与父本一致的测序深度。通过ΔSNP-index 确定父母本与混池间的位点差异[19]。
1.4 InDel 标记开发
根据大豆Williams 82 (Glycine maxV2.0)的参考基因组序列,利用SLAF-BSA 高通量测序结果,确定了调控大豆主茎节数的物理区间。然后利用本研究中119 和C025 25×深度的重测序信息,使用GATK[20]软件工具包开发目标区间的InDel 标记。
1.5 目标区间精细定位
利用新开发的InDel 标记在RIL 群体中进行基因型鉴定,结合表型分析,对目标区间精细定位。QTL 扫描采用WinQTL Cartographer 2.5 软件(http:/ /statgen.ncsu. edu/qtlcart/WQTLCart.htm)中的复合区间作图法。叶片的DNA 提取采用改良的CTAB 方法[21],PCR扩增以及PCR 产物进行8%聚丙烯酰胺凝胶电泳、显影、染色和带型判读参照文献[22-23]进行。
2 结果与分析
2.1 父母本和RIL 群体主茎节数表型变异分析
多年试验发现亲本C025 的平均主茎节数为9.13,中119 的平均主茎节数为16.59,亲本间的主茎节数相差近2 倍(表1)。本研究利用单粒传法获得含有102 个单株/株系的RIL 群体(F5),2017年和2018年两年调查RIL 群体主茎节数,发现分离群体主茎节数表现出广泛的变异(4.03~19.19)(图1)。
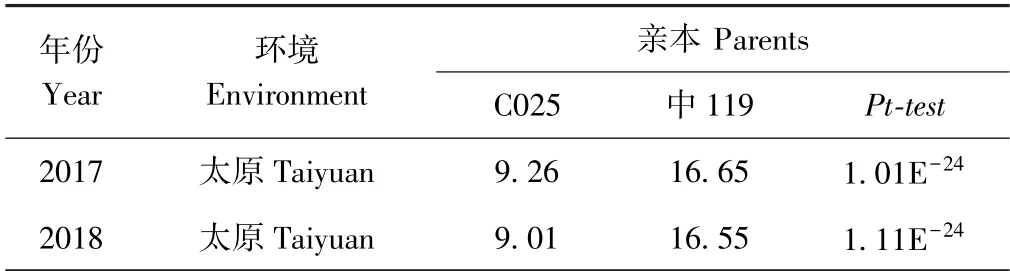
表1 双亲主茎节数表型分析Table 1 The phenotypic variation of node numbers on the main stem in parents
2.2 SLAF 测序数据分析和评估以及SALF 标签和SNP 标记的开发
利用SLAF 测序对双亲和RIL 群体多少主茎节数混池进行序列分析,限制性内切酶RsaI 和HaeIII 酶切的SLAF 片段长度在364~414 bp。然后通过BWA[24]软件将样本的测序reads 与参考基因组进行比对,结果显示本次试验双端比对效率在90.34%,说明比对效率正常。测序结果表明,Q30 的百分比平均含量为95.35%,GC 的百分比平均含量为40.27%。父本中119(aa)过滤后的reads 数为15 123 276,Q30 的百分比含量为95.71%,GC 的百分比含量为40.10%;母本C025(ab)过滤后的reads 数为17 566 874,Q30 的百分比含量为95.67%,GC 的百分比含量为39.99%;后代RIL 极端混池(ac 和ad)过滤后的reads 数分别为25 436 458 和21 960 126,Q30 的百分比含量分别为95.30%和94.70%,GC 的百分比含量分别为40.52%和40.48%(表2)。

表2 高通量测序数据挖掘表Table 2 Mining results of the high-throughput sequencing data
本研究共开发了1 003 590 个SLAF 标签,父母本的平均测序深度为28.48×,而混合池的为36.16×。其中父本中119 共获得241 118 个SLAF 标签数,平均测序深度为26.65×,母本C025 共获得240 712 个SLAF标签数,平均测序深度为30.30×。RIL 群体两个极端混池获得SLAF 标签数分别为260 003 和261 757,平均测序深度分别为38.89×和33.43×(表3)。
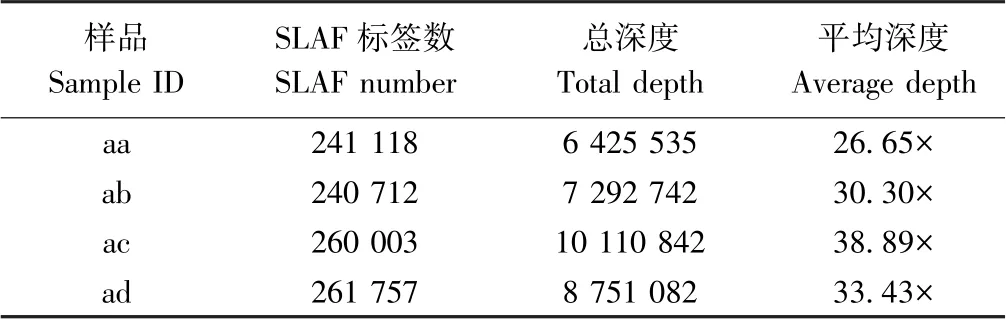
表3 测序开发的SLAF 标签数Table 3 Sequencing data of the developed SLAF markers
SNP 的检测主要使用GATK[18]软件工具包和samtools[16]进行变异检测,以确保其准确性。将以上两种方法分别得到的变异位点一致的SNP 用于后续分析。双亲和RIL 群体极端混池4 个样品的SNP 统计结果如图2 所示,共检测到193 082 个SNP 位点,父母本检测到的SNP 位点略低于两个混池,但是4 个样本间均有重复的SNP 位点。同时对不同染色体进行SLAF 标签和SNP 标记开发(图3)。
2.3 主茎节数关联分析
在关联分析前,首先对SNP 进行过滤,共过滤了166 530 个SNP,最终得到高质量的可信SNP 位点26 552 个。然后进行ED 值分析,统计各个碱基在不同混池中的深度,并计算每个位点ED 值,最后取所有位点拟合值的median+3SD =0. 23 作为分析的关联阈值[18]。根据关联阈值判定,在4 号染色体共得到28 个区间,总长度为16. 44 Mb,共包含1 287 个基因,其中非同义突变SNP 位点的基因共35 个(表4)。

表4 依据ED 值关联区间信息统计表Table 4 The information of the association region by ED value
采用SNP-index 方法关联分析前需要对SNP 位点进行过滤,获得过滤后的高质量可信26 552 个SNP 位点。利用SNPNUM 方法对ΔSNP-index 进行拟合(图4),结合群体的理论分离比得到阈值为0.36,最终在4号染色体上关联5 个区间,总长度为1.66 Mb,共包含120 个基因,其中非同义突变的基因共6 个(表5)。
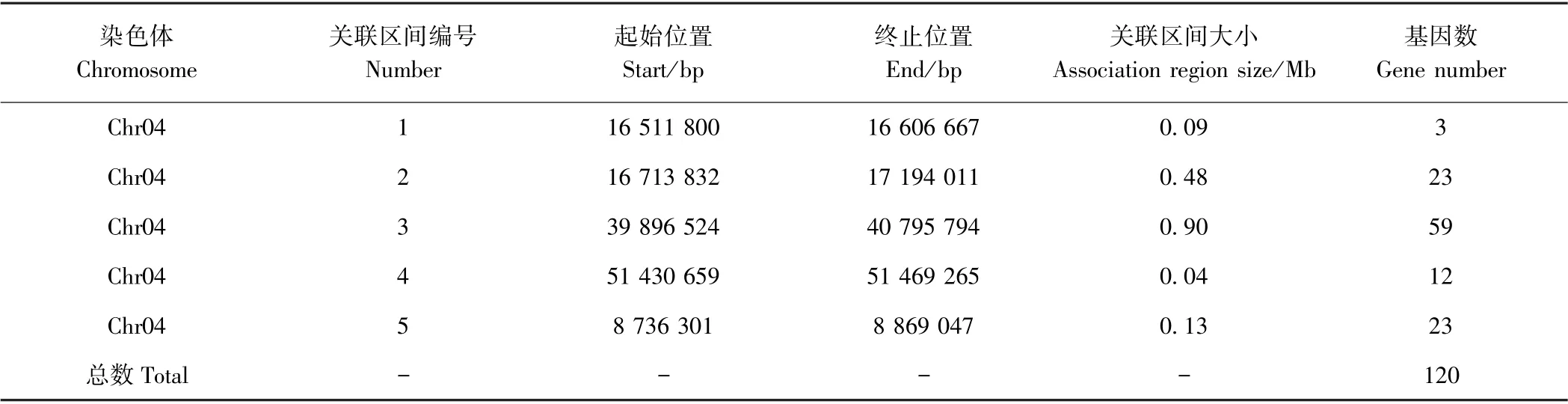
表5 依据SNP-index 值关联区间信息统计表Table 5 The information of the association region by SNP-index value
最后取两种分析方法的结果交集,结果显示ED值方法关联结果包含了SNP-index 关联分析方法,因此最终取ΔSNP-index 方法关联结果。
2.4 关联区间基因功能注释以及InDel 标记的开发
利用BLAST 软件将相关区的120 个基因与NR[24]、SwissProt、GO[25]、COG[26]和KEGG[27]数据库进行比对,最终获得113 个基因注释信息。其中KEGG中38 个基因的注释参与了核糖体、甘氨酸、丝氨酸和苏氨酸代谢、氧化磷酸化、植物激素信号转导、谷胱甘肽代谢和泛素介导的蛋白质水解等8 条信号通路。
然后利用父母本C025 和中119 重测序信息,分析1.66 Mb 关联区间双亲的基因组信息,共得到4 214 个InDel 位点。在第5 个关联候选区间进行引物设计,并将设计的引物在亲本间进行多态性筛选,最终获得了67 对InDel 引物(图5)。
2.5 关联主效区间精细定位
利用每个区间开发的新Indel 标记对双亲和F2群体进行基因型验证,结果显示在InDel 标记Chr04-21和Chr04-55 间检测到一个与大豆主茎节数相关的主效QTL(图6),同时这个位置也是关联区间的第3 个关联区间。因此初步将候选区间缩小到第3 区间0.9 Mb 范围内。
为了进一步精细定位主效区间,本研究在第3 区间利用新开发的8 个共显性InDel 标记对102 个株系的RIL 群体进行基因型鉴定,结果如图7 所示,有6 种交换类型,共9 个交换单株。第一种交换类型是在Chr04-46 标记位置发生交换,有18RIL117-3 和18RIL119-2 两个交换单株,但这2 个单株的表型与亲本C025 基本一致。第二种交换类型是在Chr04-31之前插入与亲本C025 一致片段,在Chr04-38 之后插入与亲本中119 一致片段,而在Chr04-31 和Chr04-38 之间插入的杂合片段,其中有一个交换单株18RIL11-3,但是在主茎节数表型上有一定的改变,主茎节数为12.40 鉴于双亲之间。第三种交换类型是在Chr04-38 之前插入与亲本C025 一致片段,在Chr04-46之后插入与亲本中119 一致片段,而在Chr04-38 和Chr04-46 之间插入的杂合片段,有一个交换单株18RIL65-2,主茎节数表型为14.45。第四种交换类型是在Chr04-33 之前插入与亲本C025 一致片段,之后插入与亲本中119 一致片段,有两个交换单株18RIL88-2 和18RIL87-1,主茎节数分别为15.23 和16.15。第五种交换类型是在Chr04-38 之前插入与中119 一致片段,之后插入与亲本C025 一致片段,有一个交换单株18RIL38-1,主茎节数为9.33。第六种交换类型是在Chr04-46 之前插入与中119 一致片段,之后插入与亲本C025 一致片段,有两个交换单株18RIL37-1 和18RIL44-2,主茎节数分别为16.71 和16.34。从以上六种交换类型可以看出第三、第四和第六种交换类型的主茎节数与亲本中119 接近,而第一、第二和第五种交换类型的主茎节数与亲本C025 较接近。说明Chr04-38 标记位点之前插入双亲一致片段表型不会发生变化,Chr04-46 标记位点之前插入双亲一致片段表型也不会发生变化,而在Chr04-38 和Chr04-46 标记之间插入亲本中119 或者杂合片段的交换单株表型都会接近中119。因此可以确定将与大豆主茎节数关联位点定位于Chr04-38 和Chr04-46 标记之间,物理位置为171.9 kb,包含候选基因6 个(表6)。

表6 关联区间6 个候选基因的注释信息Table 6 Annotation information of 6 genes in related interval
3 讨论
前人对农作物农艺性状的定位研究,大多数利用的是传统遗传图谱定位方法,从而确定与农艺性状紧密连锁的分子标记[28]。本研究利用的BSA 方法是针对具有明显极端分化的农艺性状,是一种快速鉴定与目标基因(区间)连锁分子标记的有效方法[29]。但是用BSA 方法结合常规遗传定位会受到DNA 分子(SSR标记等)标记数量少的限制[19],低密度的标记会影响结果的准确性。然而高通量测序的发展,为揭示基因组的遗传多样性和加速基因定位以及分离提供了一种全面实惠的手段。本研究将SLAF-Seq 高通量测序和BSA 方法有效的结合突破了DNA 标记有限的瓶颈,并且也不需要将群体全部基因型分型。目前该手段已经应用于小麦[30]、水稻[31]、高粱[32]和向日葵[33]等多种植物。本研究利用BSA-SLAF-Seq 方法在大豆中共开发了1 003 590 个SLAF 标签,鉴定出193 082 个SNP标记,均具有较高的数量和质量,并且分布在大豆每条染色体上(图3)。经过过滤后得到可信SNP 位点26 552 个,利用这些SNP 位点关联分析与大豆主茎节数相关区间。与目前开发的SSR 标记相比,本研究开发的SNP 位点为后续关联分析提供了足够的数据。说明SLAF-Seq 技术是一种高效、高分辨率的定位技术,具有成功率高、特异性强、稳定性好、成本效益高等特点。SLAF-Seq 技术与BSA 的结合为鉴定主茎节数相关的基因组区间提供了一种有效的方法。
近年来,分子标记在许多作物育种中得到了广泛的应用[34]。与传统育种方法相比,分子标记辅助育种能够提高育种效率,加快育种进程。因此,开发与目标基因(基因区间)紧密连锁的标记对育种具有重要意义。InDel 分子标记被认为是一种适应性强、高效、稳定且成本低易于普及应用的标记。本研究利用BSASLAF-Seq 方法确定了在4 号染色体上与大豆主茎节数相关的5 个区间,总长度为1.66 Mb,且该区间包含120 个基因(表5)。依据双亲的重测序信息,开发InDel 分子标记并加密候选关联区间,并精细定位大豆主茎节数。首先利用关联区间新开发的InDel 分子标记在F2群体扫描与主茎节数相关的QTL,结果在第三关联区间检测到一个峰值相关性较高的与主茎节数相关的QTL 位点,由最初的1.66 Mb 缩小到0.45 Mb。为了更进一步精细定位主效区间,本研究利用主效区间新开发的Indel 标记,进一步通过对RIL 群体全部株系进行基因分型,最终筛选到9 个交换单株/系,将主效区间分为6 种交换类型,将大豆主茎节数精细定位到InDel 标记Chr04-38 和Chr04-46 之间,其候选区间只有171.9 kb,包含6 个基因(表6)。然而将本研究结果与前人通过连锁定位研究主茎节数QTL 位点相比较发现,4 号、6 号和19 号染色体与大豆主茎节数和株高相关的QTL 位点较多[35],且Chang 等[36]研究发现大豆主茎节数与大豆分枝数存在显著的正相关,因此同一个QTL 位点有可能存在多效性。如Li等[37]研究发现大豆株高QTL-qPH-C1-3 和主茎节数QTL- qMS-C1-1 同时位于4 号染色体(50.35~52.38 Mb)且位置有部分重叠,并且这个位置与本研究主效位点比较接近,该位点在多个研究中都有重叠,说明4号染色体末端是一个调控大豆生长和发育的热点[38]。但是不同研究同样提出大豆主茎节数和株高的遗传机制也存在差异,有些位点单独调控株高,而有些位点单独调控主茎节数,说明调控大豆主茎节数和株高的遗传机制相似但又存在一定差异[37]。
4 结论
本研究利用SLAF-Seq 技术与BSA 结合的方法鉴定与大豆主茎节数紧密关联的区间,同时利用关联区间的InDel 标记将与大豆主茎节数位点精细定位到171.9 kb 区间内,包含6 个候选基因,实现了大豆主茎节数的主效位点精细定位。本研究开发InDel 分子标记Chr04-38 和Chr04-46 是与大豆主茎节数紧密相关,这两个标记为大豆主茎节数分子标记辅助育种以及后期的功能评估具有一定的应用价值。后期对该区间6 个候选基因进一步深入研究,将为大豆主茎节数基因克隆以及功能研究奠定分子基础。
猜你喜欢
中学数学研究(广东)(2023年9期)2023-06-03 03:32:40
安徽农业科学(2022年19期)2022-10-29 08:55:54
安徽农业大学学报(2022年3期)2022-10-25 12:32:36
中学生数理化·八年级物理人教版(2022年9期)2022-10-24 07:03:48
种子(2021年7期)2021-08-19 01:46:58
河北科技师范学院学报(2020年1期)2020-07-02 03:37:22
作文周刊(高考版)(2016年16期)2017-06-01 15:41:00
中国马铃薯(2017年1期)2017-03-02 09:15:46
党建文汇·下(2016年5期)2016-05-14 11:34:41
奥秘(2016年4期)2016-04-21 17:31:28
