
Toll样受体与心血管疾病关系的研究进展
2021-09-13 07:17邢云邓伟唐其柱
心血管病学进展 2021年8期
邢云 邓伟 唐其柱
(武汉大学人民医院心血管内科 代谢与相关慢病湖北省重点实验室,湖北 武汉 430060)
Toll样受体(Toll-like receptors,TLRs)是最早发现于果蝇的一类模式识别受体[1],在果蝇胚胎发育过程中,dToll基因决定着果蝇的背腹侧分化,其编码的蛋向质称为Toll样蛋白,参与果蝇的免疫反应。Medzhitov等[2]首先发现与dToll同源的人hToll基因及其编码的Toll样蛋白,并将Toll样蛋白命名为TLRs。
1 TLRs概述
1.1 TLRs的发现与分布
TLRs是一种Ⅰ型跨膜糖蛋白,包括富含亮氨酸重复序列的膜外区(识别并结合病原相关分子)、跨膜区和含有Toll/白介素(interleukin,IL)-1受体同源区结构域(Toll/IL-1 receptor domain,TIR结构域)的胞质区(介导下游信号转导)。在质膜上表达的TLRs识别病原体细胞外区的成分,包括脂蛋白(TLR1、2和6)、脂多糖(lipopolysaccharide,LPS)(TLR4)和细菌鞭毛蛋白(TLR5)。在内小体室中表达的TLRs识别病原体细胞内室中的成分,包括双链RNA(dsRNA)TLR3,单链RNA(ssRNA)TLR7和TLR8以及非甲基化胞嘧啶-磷酸-鸟嘌呤(CpG)-DNA(TLR9)[3]。目前,在哺乳动物体中已发现13种TLRs,其中人体内发现10种,小鼠体内发现12种[4]。TLRs主要分布在淋巴细胞、白细胞和单核巨噬细胞等免疫细胞表面,在非淋巴组织中也有不同程度的表达[5]。见表1。
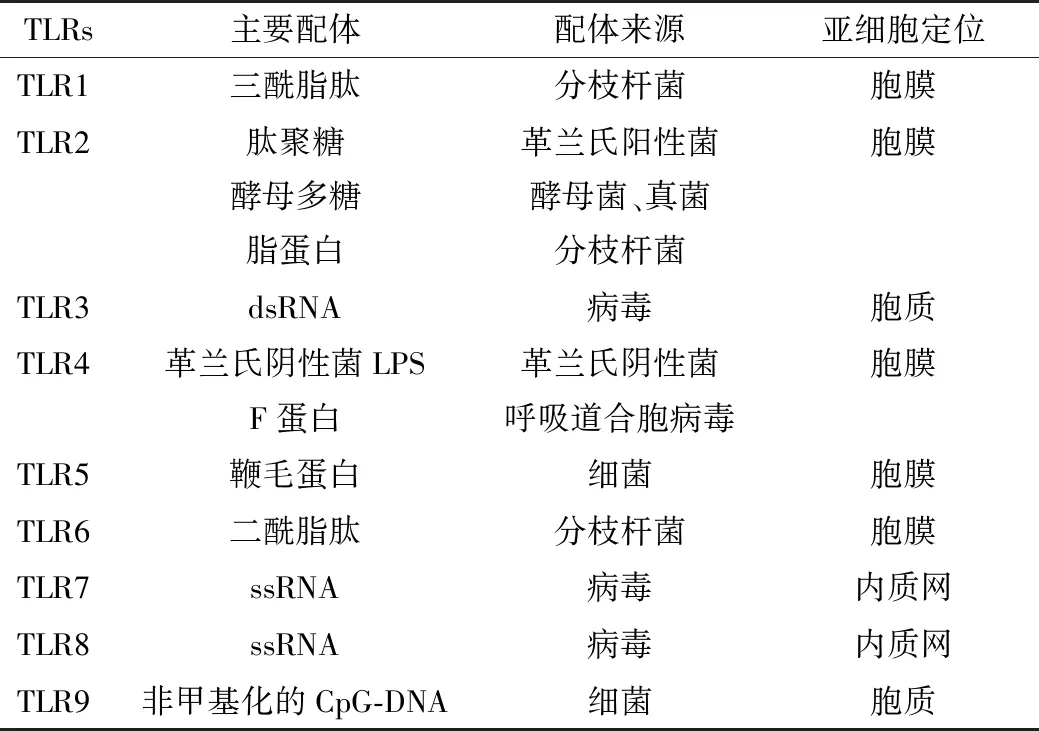
表1 TLRs的激活和分布
1.2 TLRs有关的信号通路
TLRs的胞浆区与白介素-1受体(IL-1R)家族成员胞浆区高度同源,即TIR结构域。TIR是TLRs向下游传导信号的核心部位,目前已鉴定出5种含有TIR结构域的配体,其中包含髓样分化因子88(MyD88)、MyD88配体样蛋白和含有TIR结构域的配体蛋白诱导β干扰素产生的转录因子(TRIF)、TRIF相关配体分子(TRAM)和一种含基序蛋白[6]。TLR信号可分为两种通路,即MyD88依赖通路和依赖于TRIF的信号通路。仅TLR3依赖于TRIF的信号通路,而TLR4则二者均可触发[7]。
TLRs通过多种识别分子与其配体结合[8]后,通过依赖于MyD88的信号通路和依赖于TRIF的信号通路,激活核因子κB(nuclear factor-κB,NF-κB)、干扰素调节因子(interferon regulatory factor,IRF)、丝裂原激活的蛋白激酶(mitogen-activated protein kinase,MAPK)p38和Jun激酶等[9],诱导免疫相关基因的表达。且TLR4与髓样分化蛋白-2(MD-2)和CD14能协同识别可导致机体败血症和感染性休克的革兰氏阴性菌的LPS[10]。下面以TLR4为例介绍两种传导途径。
1.2.1 依赖于MyD88的信号通路
以MyD88、IL-1受体相关激酶(IRAK)[4]等复合物的形成为开始,以NF-κB和MAPK的早期活化为特征。TLR4与配体结合后通过TIR结构域与MyD88羧基端的TIR结构域相互作用后,经过一系列过程,使磷酸化的IRAK与肿瘤坏死因子受体相关因子(TRAF)6结合[11],而后通过适配体蛋白激活转化生长因子-β活化激酶1,最终NF-κB抑制蛋白的活化导致转录因子NF-κB的激活和转位[6]。同时,TRAF6还可结合Toll途经中进化保守的信号转导中间体将TRAF6与MAPK通路联合起来,激活Jun激酶和p38 MAPK通路[12],最终导致相关基因的转录。见图1。
1.2.2 依赖于TRIF的信号通路
依赖于TRIF的信号通路通过TRIF和TRAF3激活[13],导致IκB激酶/TANK结合激酶1(IKKε/TBK1)的募集、IRF3磷酸化以及β干扰素的表达。依赖于TRIF的信号通路主要负责LPS诱导的干扰素诱导蛋白10(IP-10)、糖皮质激素终止反应基因16(GARG-16)、IRF1表达[14]以及树突状细胞成熟。见图1。
2 TLRs与心血管疾病
TLRs参与炎症和免疫反应的激活,是先天免疫的第一道防线。TLRs在各种病理条件下发挥着广泛的作用,包括心血管疾病、过敏性疾病、肥胖相关代谢性疾病、神经元变性、自身免疫性疾病、传染病和炎症性肠病。其中,TLRs在心肌炎症信号通路中发挥着关键作用,包括动脉粥样硬化(atherosclerosis,AS)、心肌梗死(myocardial infarction,MI)、心肌缺血再灌注损伤(myocardial ischemia reperfusion injury,MIRI)和病毒性心肌炎(viral myocarditis,VMC)等[15]。接下来就TLRs与相关心血管疾病的关系展开介绍。
2.1 TLRs与AS
AS进展过程中,心肌细胞出现巨噬细胞和T细胞的广泛浸润,炎症成分与斑块破裂有关,可导致MI或卒中。在AS的发生和发展过程中,病变部位的血管细胞表达多个TLRs,表明这些TLRs可能是影响AS的关键因素。
目前,已证实TLRs参与微生物感染后AS进展的信号通路。有研究表明,TLR2和TLR4在肺炎链球菌感染后激活巨噬细胞和内皮细胞,抑制胆固醇外流和促进泡沫细胞形成方面起着重要作用[16]。活化的血小板通过TLR4信号通路促进血小板微粒的分泌,且二者相互作用可产生很强的促凝功能,因此特异性阻断TLR4可防止血小板微粒与血小板相互作用,且可能成为未来抗血栓治疗的潜在靶点[17]。研究指出,TLR7在T细胞、巨噬细胞以及毛细血管内皮细胞中表达,TLR7转录水平与不良心血管事件的发生相关。实验发现TLR7可通过抑制炎症因子的表达调节AS中的炎症反应,因此TLR7可能成为AS预后的重要标志物[18]。另外,TLR9基因敲除小鼠的粥样硬化斑块中的脂质沉积和巨噬细胞数量比非基因敲除小鼠大大减少,这表明TLR9在血管炎症和AS的发展中起着关键作用[19]。在病变的动脉外膜,成纤维细胞可表达TLR4,并可在LPS的刺激下合成大量的炎性细胞因子,包括促进T细胞表达与分泌调节因子、γ干扰素诱导蛋白-10(interferon γ-inducible protein-10,IP-10)、巨噬细胞炎症蛋白和IL-8等炎性因子[20]。这些因子均被证实与AS的发生和发展有关。因此,TLRs的激活可能为AS的治疗提供新思路。
2.2 TLRs与MI
MI患者预后不良的部分原因是心肌细胞大量凋亡、坏死,心脏愈合和血管生成受限以及心功能不全,免疫细胞功能障碍导致MI后伤口不愈合或愈合不良。TLRs作为天然免疫系统的重要组成部分,在调节心肌细胞存活和伤口愈合中起着至关重要的作用。在MI动物模型中,干扰NLRP3炎性小体、TLR2和TLR4可使梗死面积减小,并可改善MI后心功能[21],因此NLRP3炎性小体、TLR2和TLR4炎症反应信号可能是临床治疗的潜在干预靶点。有研究发现敲除TLR7基因可抑制心肌缺血后的炎症进程,同时促进小鼠MI后的心肌细胞存活,并可减少左心室重塑[22]。同时,本研究团队揭示了高迁移率族蛋白A1(HMGA1)和TLR9的相互作用可促进心肌细胞的存活、伤口愈合和MI后血管生成[23]。另有研究表明,细胞衍生因子-1能减少MI患者中心肌细胞的凋亡,促进血管再生,改善心功能,进一步研究发现这与TLR4/NF-κB信号通路的激活有关[24]。有实验者用TLR3配体对新生心肌细胞进行体外处理,结果发现细胞糖酵解代谢和心肌细胞增殖得到显著增强,因此TLR3可能是MI后心脏再生和修复所必需的[25]。
因此,TLRs与不同病因所致MI相关,尽管MI后短期给药对减少心脏损伤和不良心血管事件的发生作用有限,但考虑到炎症因素对缺血性心脏病的长期影响,发病后针对TLRs的抗炎症治疗仍可取得较大的收益。
2.3 TLRs与MIRI
缺血和再灌注改变细胞内环境的氧化还原状态,诱导大量促炎因子释放,后者使TLRs激活并促进中性粒细胞释放活性氧/活性氮和蛋白水解酶[26],从而导致大量炎性细胞浸润和氧化应激反应,加剧组织损伤[27-28]。研究发现外源性RNA可诱导巨噬细胞和中性粒细胞产生巨噬细胞炎症蛋白-2、肿瘤坏死因子α等炎性因子,而在TLR7基因缺陷的细胞中,RNA诱导产生细胞因子的功能被部分抑制,表明外源性RNA可通过TLR7活化的信号通路诱导细胞炎症因子的产生[29]。
细胞凋亡在MIRI中起着重要作用,MIRI时引起的细胞凋亡由氧化应激、细胞内钙超载、酸中毒以及坏死细胞分解释放的细胞毒素引起的炎症反应所致,尤其是再灌注时产生大量活性氧导致脂质过氧化,蛋白质和酶分子失活。最近的研究发现,损伤细胞可通过释放RNA激活TLR3,进而促进心脏移植后MIRI的发生和发展,揭示了TLR3在再灌注引起的炎症损伤中的作用[30]。通常认为TLR2、TLR3和TLR4的激活在MI中有害,然而近来发现TLR5在限制心肌损伤、炎症激活和预防心肌功能损害方面发挥了有利作用[31]。另外,阻断TLR9介导的信号通路可减轻炎症,降解梗死释放的细胞外线粒体DNA,减轻MIRI,因此TLR9可作为MIRI的治疗靶点[32]。这些发现为TLRs作为未来药物治疗靶点和防止MIRI的发生提供了思路。
2.4 TLRs与VMC
VMC常由肠道病毒、腺病毒、人类细小病毒B19、人类疱疹病毒6型和柯萨奇病毒B组(CVB)引起,其中CVB等嗜心性病毒引起的直接损伤及其所诱发的免疫反应是心脏损害的主要原因,具体机制可能是病毒引起炎性细胞大量释放,炎性细胞识别CVB3并感染触发NF-κB激活,分泌IL-6、IL-1β、肿瘤坏死因子α和IL-8等炎性因子,从而引起炎症损伤[33]。研究表明,Th1(以介导细胞免疫反应为主)型免疫通过减少病毒复制来减轻急性心肌炎的临床症状,并通过抑制Th2(以介导体液免疫反应为主)反应来阻止慢性心肌炎和扩张型心肌病的进展,而Th2型免疫反应通过调节性T细胞和抗炎细胞因子抑制Th1反应来减轻急性心肌炎[34]。不同的病理状态下,Th1和Th2免疫反应的升高对于从心肌炎到扩张型心肌病和心力衰竭的进展至关重要。
近期,Tatsumi等[35]以CVB3或甲型H1N1流感病毒感染小鼠来源的心肌细胞,发现刺激蛋白酶激活受体4(PAR4)可增强TLR3依赖的趋化因子10的表达。且PAR4基因缺陷小鼠的抗病毒作用被抑制,表现为CVB3介导的VMC恶化,死亡率增加。此研究表明,PAR4具有作为干预靶点减轻CVB3和甲型H1N1流感病毒感染导致的心肌损伤的潜在优势。此外,有研究者发现中药黄芪中的黄芪多糖通过抑制TLR4/NF-κB p65信号通路的激活而减轻炎症反应,改善CVB3诱导的VMC[36]。另外,MyD88基因敲除可抑制病毒感染介导的促炎因子(如IL-1β)分泌,同时提高心肌细胞生存率[37]。
此外,非微生物因素也可通过TLRs激活参与炎症反应。例如,坏死细胞RNA通过TLR7-MyD88信号诱导细胞因子的产生引发心肌炎[38]。体外血流动力学应激损伤的线粒体被心肌细胞中的自噬/溶酶体系统降解后将导致TLR9介导的心肌细胞炎症反应,并能诱导心肌炎和扩张型心肌病[39]。
以上证据表明,TLRs在CVB3甚至非微生物因素诱导的VMC中起着重要作用,且TLRs基因在严重心力衰竭患者心脏组织中的表达增加[40]。因此抑制TLRs可能为临床治疗VMC提供新方法。
3 总结与展望
综上所述,TLRs在AS、MI、MIRI和VMC等心血管疾病的发生和发展过程中发挥着重要作用。TLRs通过MyD88依赖通路和TRIF依赖通路促进大量炎症因子、细胞表面分子和化学因子的分泌,进一步加剧了心肌的损伤。目前,以TLRs为靶点的药物研究日益受到重视,通过干预、调节抑制TLRs的表达可望成为一种心血管疾病防治的新手段。
猜你喜欢
中西医结合心脑血管病杂志(2022年19期)2022-11-19
中国交通信息化(2022年8期)2022-10-28
中国种业(2022年9期)2022-10-13
中国现代医生(2022年21期)2022-08-22
九江学院学报(自然科学版)(2022年2期)2022-07-02
中西医结合心脑血管病杂志(2022年4期)2022-03-11
中西医结合心脑血管病杂志(2022年2期)2022-02-15
昆明医科大学学报(2021年12期)2021-12-30
现代临床医学(2021年1期)2021-01-26
体育科学(2018年12期)2019-01-04
