
亚临界水中葡萄糖转化生成5-羟甲基糠醛的理论研究
2021-09-10 07:08阎智锋何春启岳秀萍王玉萍卢建军
燃料化学学报 2021年8期
阎智锋,连 洁,赵 舟,何春启,岳秀萍,王玉萍,吴 杏,卢建军,5
(1.太原理工大学 轻纺工程学院,山西 太原 030024;2.太原理工大学 环境科学与工程学院,山西 太原 030024;3.国家先进功能纤维创新中心,江苏 苏州 215228;4.西安工程大学 纺织科学与工程学院,陕西 西安 710048;5.太原理工大学 煤科学与技术教育部与山西省重点实验室,山西 太原 030024)
随着化石能源的日益消耗和环境问题的日益严重,生物质资源作为唯一的可再生碳源,被期望得以资源化利用并建立可持续的化学工业,从而降低人类对于化石能源的依赖[1]。5-羟甲基糠醛(5-HMF)是重要的生物质平台化合物,可由葡萄糖或果糖脱水制得[2,3]。当反应物为果糖时,其反应性和目标产物5-HMF 的选择性均高于葡萄糖转化生成5-HMF 的反应[4]。然而果糖原料成本较高,而葡萄糖作为纤维素的基本结构单元,产量丰富,廉价易得,用其替代果糖制备5-HMF 具有更大的研究价值。目前,多数研究认为果糖是葡萄糖转化生成5-HMF 的关键中间体,即葡萄糖首先异构化生成果糖,然后果糖经三次连续脱水生成5-HMF[4-6],如图1 所示。

图1 葡萄糖脱水制备5-HMF 反应机理Figure 1 Reaction mechanism for dehydration of glucose to 5-HMF
酸和碱均可催化葡萄糖异构化反应[7-11],然而由于葡萄糖中多羟基与C-C 骨架结构的相似性,不同类型的催化剂具有不同的劣势:Brønsted 碱催化剂极易引发其他副反应的发生,导致果糖选择性较低[7];Brønsted 酸则对该异构化反应的催化效率较低;Lewis 酸能够高效地催化该异构化反应,但反应时间较长且葡萄糖的强极性易导致Lewis酸催化剂水解失活[9]。有研究认为,Brønsted 酸碱与Lewis 酸催化作用的差异可能源于催化机理的不同[8]。Brønsted 酸碱催化作用下葡萄糖异构化为果糖的反应为质子转移过程,即吡喃葡萄糖开环后发生LBAE 转化(Lobry de Bruyn-Albeda van Ekenstein)生成开环果糖,然后闭环形成呋喃果糖。而Lewis酸催化作用下为氢转移过程,即Lewis 酸中心与吡喃葡萄糖的羰基和α-羟基形成环状螯合物,然后C(2)的氢直接向C(1)转移生成开环果糖[8]。与质子转移过程相比,环状螯合物的形成明显降低了氢转移过程所需活化能且对果糖选择性较高,因而催化效果较好。果糖脱水制备5-HMF 的反应则被广泛认为是Brønsted 酸催化过程,即质子催化作用下呋喃果糖依次脱除三分子水。
由此可见,葡萄糖异构化为果糖需要碱性活性中心或Lewis 酸性中心的催化,而果糖继续脱水生成5-HMF 需要Brønsted 酸性中心催化。这表明在同一催化体系下实现葡萄糖到5-HMF 的高效定向转化,需要对催化体系的催化活性中心类型及溶剂进行进一步优化设计。目前的研究多集中于高效催化活性中心的结构调控及其催化性能调变,而对于溶剂筛选及溶剂在反应中作用的研究报道较少。实际上,通过选择合适类型的溶剂并调控溶剂的组成与配比,可以实现对反应选择性和转化率的优化[12]。
目前,溶剂的筛选主要是基于经验主义的试错法。溶剂对反应的作用主要包括溶剂化效应与催化作用两点。溶剂化效应,即溶剂与溶质之间产生的分子间作用力,改变了反应物和过渡态的稳定性,进而影响反应活化能和反应速率。如三聚氯氰催化作用下酮肟的贝克曼重排反应,当溶剂为六氟异丙醇时,少量三聚氯氰即可在室温下较短时间内得到高产率的酰胺产物,优于二甲基甲酰胺、二氯乙烷、四氢呋喃作溶剂时的反应条件和产率[3,13]。更重要的是,溶剂很可能直接参与反应,起到催化剂的作用,从而加速反应的发生[14]。然而,由于分析手段的局限性,实验结果并不能完全准确描述溶剂在化学反应过程的作用,溶剂对化学反应的影响规律和作用机理还缺乏深入的理解。
由于量子化学方法在洞悉微观机理方面的优势,近年来被用于研究多种反应体系的微观机理,对于描述液相体系中的溶剂效应也得到了较大的关注。液相反应体系的描述主要包括隐性溶剂模型和显性溶剂模型两种。隐性溶剂模型如极化连续介质模型(polarizable continuum model,PCM)、溶剂密度模型(solvent model density,SMD)、似导体屏蔽模型(conductor-lick screening model,COSMO),即将溶剂视为连续介质,并通过介电常数来描述不同溶剂的体相性质。显性溶剂模型则是在通过介电常数描述体相溶剂效应的基础上,采用显性溶剂分子描述微观溶剂化作用,实际上是极化连续介质模型与微观溶剂化相结合的一种杂化算法。与隐性溶剂相比,显性溶剂模型在短程作用、溶剂取向、离子分布和非物理作用等方面更接近真实情况,因此,一些基于隐性溶剂模型的计算结果与显性溶剂模型的计算结果并不吻合,与实验值的结果更是相差甚远。Shigemoto 等[15]以极化连续介质模型研究了涤纶的非催化水热降解,结果表明其反应活化能为48.0-51.1 kcal/mol,但是实验数据和动力学研究则表明该反应的水热降解活化能为21.5-35.85 kcal/mol[16-21]。本课题组[22]前期采用色散修正密度泛函理论(dispersion-corrected density functional theory,DFT-D)研究了显性溶剂模型描述的涤棉水热分离机理,结果表明显性溶剂水分子不但对酯基和糖苷键具有诱导活化作用,而且确实可以参与到反应中协助质子的迁移,缩短质子迁移的距离,从而降低过渡态的自由能及相应活化能,促进反应的进行。因此,显性溶剂模型可以更为准确地描述溶剂化效应对几何结构、反应机理及其能量变化等方面的影响。但是由于显性溶剂模型的计算量较大,目前隐性溶剂模型仍然是主流[23,24]。
近年来,环境问题引起全球范围的日益关注,超/亚临界水由于其无毒、环保、经济等特点,且有低相对介电常数、高离子积、低黏度和表面张力等诸多理化特性,重新受到了研究者的重视。目前酸碱双功能催化剂绝大部分都是固体,而超/亚临界水是一种具有较高浓度的H+和OH-的中性液体溶剂[25],对反应具有独特的酸碱双功能催化效果,洞悉该体系下的酸碱催化反应机理及反应的关键控速步骤,对于反应体系溶剂筛选及催化剂结构设计具有重要的理论指导意义,极具研究价值。
超临界和亚临界水中生物质水解转化行为表现出一定的差异性。赵岩等[26,27]比较研究了超临界和亚临界条件下纤维素的水解过程,结果表明超临界条件下纤维素及低聚糖的水解速率小于单糖的水解速率,不利于单糖的积累;而亚临界条件下纤维素及低聚糖的水解速率大于单糖的水解速率,有利于单糖的积累。Promdej 等[28]进一步研究了不同条件下微晶纤维素水解产物分布,其实验和动力学数据显示在573 K 时果糖生成5-HMF 的速率高于葡萄糖和果糖降解生成其他液相产物的速率;当温度升至623 K 达到近临界温度时,果糖生成5-HMF 的速率与葡萄糖降解生成其他液相产物的速率相接近;当温度继续升高至超临界温度达到673 K 以上时,葡萄糖降解生成其他液相产物的速率为果糖生成5-HMF 速率的数十倍,表明亚临界温度下更有利于5-HMF 的生成,其结果与Sasaki 等[29]超/亚临界条件下纤维素水解产物分布相一致。超/亚临界条件下水解转化行为的差异性或许可以用反应类型的差异来解释[30,31],即在较高温度的超临界条件下自由基型反应更有利,而在较低温度的亚临界条件下离子型反应则更容易发生。
因此,本研究借助DFT-D 方法[32],分别通过隐性溶剂模型和显性溶剂模型,研究了亚临界水中葡萄糖异构化为果糖及果糖进一步脱水制备5-HMF的反应机理,探究显性H2O 分子参与下反应机理的准确描述,分析显性溶剂模型下过渡态结构,进而揭示H+、OH-对基元反应促进或抑制的根本原因。本研究最终形成纤维素资源化利用的理论基础,促进葡萄糖及纤维素的进一步有效利用。
1 计算模型和方法
本研究所有计算均使用Materials Studio 软件中的DMol3模块[33,34]。DFT-D 已被证明可用于评价亚临界水中的非共价作用与热力学研究[35,36],因此本研究采用DFT-D 方法[32]研究隐性溶剂模型与显性溶剂模型。隐性溶剂模型通过DMol3模块中的COSMO 方法,以介电常数ε=27 描述523.15 K、25 MPa 时亚临界水的体相溶剂效应[31,37]。显性溶剂模型的表达包括COSMO 描述的体相溶剂化效应和显性溶剂H2O 分子描述的微观溶剂化效应两部分。另外,由于H2O 为流动相,因此,当显性溶剂模型中反应中间体构型保持不变仅H2O 分子位置发生变化时,体系能量发生变化而不存在过渡态。计算过程中所有的过渡态结构均使用完全线性同步/二次线性同步(complete linear/quadratic synchronous transit method,LST/QST)的TS Search和TS Optimization 进行搜寻[38,39],并通过振动频率分析确认其有且仅有唯一虚频。
交换相关泛函采用广义梯度近似(generalized gradient approximation,Perdew-Burke-Ernzerhof,GGA-PBE)[40];所有原子采用全电子赝势进行处理且价电子波函通过双数值加极化基组DNP 进行展开。计算加以自旋非限制描述开壳层。计算过程中自洽场能量、最大力场、最大原子位移分别为2.0×10-5Ha、4.0×10-3Ha/Å和5.0×10-3Å。
2 结果与讨论
2.1 隐性溶剂模型中葡萄糖异构化为果糖反应
图2、图3、图4 分别为隐性溶剂模型中葡萄糖异构化为果糖的反应路径、对应的反应物、中间体、过渡态、产物的优化构型以及反应势能图。吡喃葡萄糖IG 首先发生分子内H 迁移,生成开环葡萄糖IGF1,过渡态TS1 为O(5) 与H 趋于成键的同时C(1)-O(5) 发生断裂,所需活化能为48.00 kcal/mol。开环葡萄糖IGF1 继续发生醛-烯醇异构化,H 由C(2)迁移至O(1),生成1,2-烯二醇中间体IGF2,该反应所需活化能为74.05 kcal/mol。然 后,IGF2 中 O(1) 与O(2) 的O-H 朝 向发生变化,分子内氢键改变,生成的IGF3 更有利于下一步的分子内H 迁移,该步基元反应不需要活化能。IGF3 继续发生烯醇-酮异构化,H 由O(2)迁移至C(1),生成酮式开环果糖IGF4,所需活化能为66.20 kcal/mol。最后,开环果糖IGF4 发生分子内H 迁移,H 由O(5)迁移至O(2)生成产物呋喃果糖IF,所需活化能为29.93 kcal/mol。图4 表明,该反应共吸收4.08 kcal/mol 能量,控速步骤为开环葡萄糖的醛-烯醇异构化IGF1→IGF2,所需活化能高达74.05 kcal/mol。

图2 隐性溶剂模型中葡萄糖异构化为果糖反应路径Figure 2 Reaction pathways for the isomerization of glucose to fructose in the implicit solvent model

图3 隐性溶剂模型中葡萄糖异构化为果糖反应路径中反应物、中间体、过渡态和产物的优化构型(Å)Figure 3 Optimized configurations of reactant,intermediates,transition states and product along the isomerization of glucose to fructose in the implicit solvent model;the bond lengths are given in Å
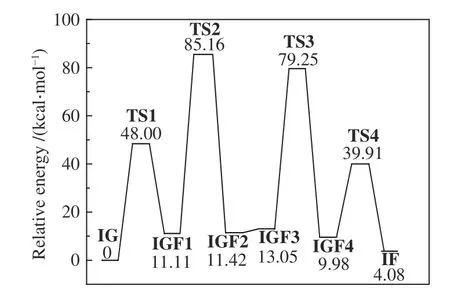
图4 隐性溶剂模型中葡萄糖异构化为果糖反应势能Figure 4 Potential energy profile along the isomerization course of glucose to fructose in the implicit solvent model
2.2 显性溶剂模型中葡萄糖异构化为果糖反应
图5、图6、图7 分别为显性溶剂模型中葡萄糖异构化为果糖的反应路径、对应的反应物、中间体、过渡态、产物的优化构型以及反应势能图。与隐性溶剂模型相比,三个显性水分子被添加至吡喃葡萄糖的O(1)H 和O(2)H 周围并与吡喃葡萄糖以分子间氢键相作用,初始优化构型如图6中EG 所示。
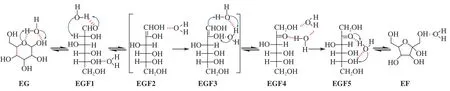
图5 显性溶剂模型中葡萄糖异构化为果糖反应路径Figure 5 Reaction pathways of the isomerization of glucose to fructose in the explicit solvent model

图6 显性溶剂模型中葡萄糖异构化为果糖反应路径中反应物、中间体、过渡态和产物的优化构型(Å)Figure 6 Optimized configurations of reactant,intermediates,transition states and product along the isomerization of glucose to fructose in the explicit solvent model;the bond lengths are given in Å
吡喃葡萄糖EG 生成开环葡萄糖EGF1 过程中,O(1)H 断裂形成的质子迁移至附近H2O 分子形成H3O+,同时H3O+中另一个质子趋于解离并与O(5)趋于成键形成过渡态TS1。由于H2O 分子的参与有利于基元反应中质子迁移,反应所需活化能明显降低,为21.65 kcal/mol。与隐性溶剂模型的分子内H 迁移相比,显性溶剂模型中过渡态TS1 的C(1)-O(5) 键长由1.607 Å增 长至1.743 Å。随后,开环葡萄糖EG1 在另一个溶剂H2O 分子协助下发生醛-烯醇异构化。C(2)-H 断裂后形成的质子向H2O 分子迁移形成过渡态TS2,此时O(H2)···H 和H···C(2)的距离分别为1.349和1.431 Å。过渡态TS2 的构型表明,开环葡萄糖发生醛-烯醇异构化的过渡态能量决定于C(2)位α-H 的提取,C(2)提取的质子很容易与H2O 结合形成H3O+并重新解离形成O(1)-H,生成开环果糖EGF2。溶剂H2O 分子的参与明显降低了该基元反应活化能,为33.89 kcal/mol。EGF2 中溶剂H2O 分子位置调整形成EGF3,以利于下一步基元反应的进行。然后,EGF3 中O(2)-H 断裂,在两个溶剂H2O 分子协助下生成的H3O+趋于解离并趋于与C(1)成键,形成过渡态TS3,此时O(H2)···H 和H···C(1) 的距离分别为1.242 和1.578 Å。与TS2 结构相似,过渡态TS3 的结构表明,葡萄糖羟基O(2)-H 的断裂与溶剂间的质子传递较为容易,其过渡态能量决定于趋于解离的H3O+与C(1) 作用结构的稳定性。烯醇-酮异构化步骤形成开环果糖EGF4,反应活化能为28.87 kcal/mol。溶剂H2O 分子位置调整后,EGF5 在H2O 协助下完成质子迁移,开环果糖EGF5 生成呋喃果糖EF。反应所需活化能为18.57 kcal/mol,过渡态结构TS4 为O(5)-H 趋于断裂形成H3O+,H3O+另一个质子趋于解离并与O(2)成键,同时C(2) 与O(5) 趋于成键。图7 表明,该反应共放出5.26 kcal/mol 能量,控速步骤为开环葡萄糖的醛-烯醇异构化EGF1→EGF2,所需活化能为33.89 kcal/mol。

图7 显性溶剂模型中葡萄糖异构化为果糖反应势能Figure 7 Potential energy profile along the isomerization course of glucose to fructose in the explicit solvent model
2.3 隐性溶剂模型中果糖脱水制备5-HMF 反应
图8、图9、图10 分别为隐性溶剂模型中果糖脱水制备5-HMF 的反应路径、对应的反应物、中间体、过渡态、产物的优化构型以及反应势能图。首先,呋喃果糖C(1)的H 与C(2)的OH 结合发生分子内脱水形成烯醇中间体IFH1,所需活化能为59.05 kcal/mol。由于IFH1 中C(1)-OH 的H与C(3)的OH 距离太远而不能直接发生分子内脱水,故本步脱水基元步骤采用隐性质子化模型。IFH1 发生质子化形成中间体IFH2,其优化构型表明IFH1 的羟基不易直接发生质子化,而是溶剂H2O 质子化形成H3O+。然后在H3O+作用下,C(3)的OH 发生质子化并脱水形成碳正离子中间体IFH3,所需活化能为9.93 kcal/mol。然后IFH3 脱质子形成IFH4,该步基元反应放出8.22 kcal/mol能 量。IFH4 中C(4) 位 的OH 与C(5) 位的H 发生第三次分子内脱水生成产物5-HMF,所需活化能为51.20 kcal/mol。图10 表明,隐性溶剂模型中果糖脱水制备5-HMF 的反应共放出14.50 kcal/mol能量,控速步骤为呋喃果糖的第一次分子内脱水即IF→IFH1,所需活化能为59.05 kcal/mol。

图8 隐性溶剂模型中果糖脱水制备5-HMF 反应路径Figure 8 Reaction pathways for the dehydration of fructose to 5-HMF in the implicit solvent model

图9 隐性溶剂模型中果糖脱水制备5-HMF 反应路径中反应物、中间体、过渡态和产物的优化构型(Å)Figure 9 Optimized configurations of reactant,intermediates,transition states and product for the dehydration of fructose to 5-HMF in the implicit solvent model;the bond lengths are given in Å

图10 隐性溶剂模型中果糖脱水制备5-HMF 反应势能Figure 10 Potential energy profile along the dehydration mechanism of fructose to 5-HMF in implicit solvent model
2.4 显性溶剂模型中果糖脱水制备5-HMF 反应
图11、图12、图13 分别为显性溶剂模型中果糖脱水制备5-HMF 的反应路径、对应的反应物、中间体、过渡态、产物的优化构型以及反应势能图。与隐性溶剂模型相比,三个显性水分子被添加至呋喃果糖的O(1)H、O(2)H 和O(4)H 周围并与呋喃果糖以分子间氢键相作用,初始优化构型如图12 中EF 所示。
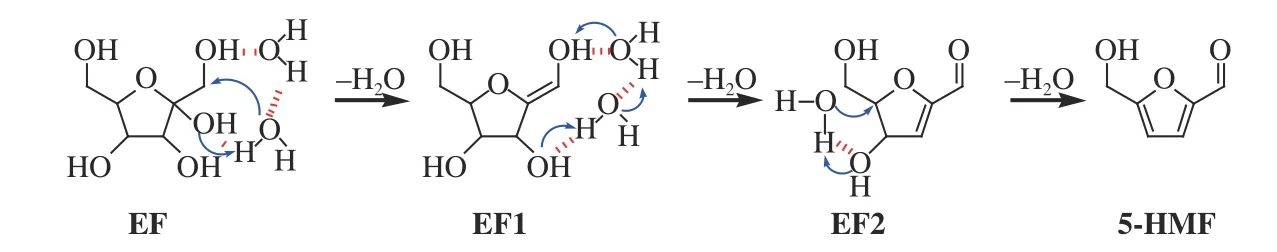
图11 显性溶剂模型中果糖脱水制备5-HMF 反应路径Figure 11 Reaction pathways for the dehydration of fructose to 5-HMF in the explicit solvent model

图12 显性溶剂模型中果糖脱水制备5-HMF 反应路径中反应物、中间体、过渡态和产物的优化构型(Å)Figure 12 Optimized configurations of reactant,intermediates,transition states and product for the dehydration of fructose to 5-HMF in the explicit solvent model;the bond lengths are given in Å
呋喃果糖EF 第一次脱水形成EF1 的过程中,溶剂H2O 分子首先发生解离,生成的质子与C(2)的OH 结合生成H2O,同时C(1) 位的H 向解离后的OH-靠近趋于成键形成过渡态TS1。由于过渡态中OH-的不稳定性,反应所需活化能虽然有所降低但仍然较高,为50.95 kcal/mol。然后,EF1 中C(1)位的OH 解离生成的质子在两个溶剂H2O 分子协助下发生质子迁移,并与C(3)位的OH 生成吸附态H2O 形成过渡态TS2,此 时O(H2)···H 和H···O(3)的距离分别为1.221 和1.230 Å。相对于隐性质子化模型,显性溶剂模型中本步基元反应所需活化能较高,为25.73 kcal/mol。TS2 中C(3)位的吸附态H2O 脱附形成EF2 并发生第三次脱水。与第一步脱水类似,溶剂H2O 分子解离生成的质子与C(4) 的OH 结合生成H2O,同时C(5)位的H 向解离后的OH-靠近趋于成键形成过渡态TS3,此 时C(5)···H 和H···OH-的距离分别为1.322 和1.425 Å,反应所需活化能为42.67 kcal/mol,比隐性溶剂模型中所需活化能略低。图13 表明,显性溶剂模型中果糖脱水制备5-HMF 共放出12.93 kcal/mol能量,控速步骤为呋喃果糖的第一次分子内脱水即EF→EF1,所需活化能为50.95 kcal/mol。

图13 显性溶剂模型中果糖脱水制备5-HMF 反应势能Figure 13 Potential energy profile along the dehydration mechanism of fructose to 5-HMF in explicit solvent model
2.5 溶剂效应分析
隐性溶剂模型与显性溶剂模型中葡萄糖异构化为果糖及果糖脱水制备5-HMF 两个反应的模拟计算表明,显性溶剂模型中溶剂H2O 分子确实能够作为质子传递型溶剂参与到反应中并在质子迁移过程起催化作用,并呈现出比隐性溶剂模型中更低的活化能,更接近真实反应的情况。然而,对于果糖脱水制备5-HMF 第二次脱水,由于隐性溶剂模型的局限性,烯醇中间体IFH1 不能直接发生分子内脱水而采用隐性质子化模型进行描述,此时该步骤反应所需活化能低于显性溶剂模型中该反应过程的描述。
进一步分析显性溶剂模型中葡萄糖异构化为果糖及果糖脱水制备5-HMF 两个反应中开环与闭环异构化、烯醇-醛酮异构化及分子内脱水三种类型的基元反应,其过渡态结构及其热力学数据表明溶剂H2O 分子的存在对不同基元反应的过渡态结构及其能量的影响作用不同。
对于开环与闭环异构化反应涉及的O-H 键的断键与O-H 键的成键,溶剂H2O 分子作为质子传递型溶剂,通过氢键作用驱动的化学键成键与断键从而降低过渡态结构的能量,显性溶剂模型中反应活化能比隐性溶剂模型中低11.36-26.35 kcal/mol。
醛-烯醇异构化反应中(EGF1→EGF2)C-H 键首先趋于断裂并形成过渡态,而烯醇-酮异构化反应中(EGF3→EGF4)中O-H 键首先发生断裂,然后C-H 键的形成决定了基元反应活化能。醛-烯醇及烯醇-酮异构化均为亲核反应并形成碳负离子过渡态结构。过渡态的结构表明,除氢键驱动力因素对过渡态结构的稳定性之外,成键断键过程中H3O+可与碳负离子在其杂化轨道方向形成较强作用力,并且碳负离子p轨道的孤对电子也能与相邻C=O 双键形成p-π共轭而降低过渡态能量,因而显性溶剂模型中反应活化能比隐性溶剂模型中低37.33-40.16 kcal/mol。醛基的α-H(EGF1)比酮基的α-H(EGF3)活化程度高,更容易脱去,但由于过渡态TS2 中醛基吸电子能力弱于TS3 中酮基吸电子能力而致使TS2 具有较高自由能,因而开环葡萄糖的醛-烯醇异构化EGF1→EGF2 为亚临界水中葡萄糖异构化为果糖的控速步骤。降低这一控速步骤活化能的直接手段在于醛式葡萄糖α-H 的提取及形成的碳负离子稳定性,这就解释了实验中Brønsted 碱催化剂容易催化葡萄糖异构制备果糖这一反应的事实。
果糖生成5-HMF 反应中包括碳链上邻位脱水和双羟基间脱水两种类型:碳链上邻位脱水为亲电反应,羟基质子化形成H2O 并脱除后形成碳正离子,然后C-H 趋于断裂形成过渡态;而双羟基间脱水则为亲核反应,羟基脱质子并通过溶剂中的质子迁移过程将另一个羟基质子化,生成吸附态H2O 而形成过渡态。邻位脱水反应的过渡态结构均存在OH-离子,由于溶剂H2O 分子数量较少而不能通过足够氢键作用将OH-的负电荷分散至较低程度,因而显性溶剂模型中反应活化能比隐性溶剂模型中低8.1-8.53 kcal/mol,相对于另外两种类型的基元反应活化能降低程度较小。两种类型脱水反应活化能相差较大,主要原因在于不同类型脱水反应的过渡态结构稳定性不同。对于碳链上邻位脱水反应,第一次脱水形成的过渡态TS1的碳正离子中C(2)位空轨道与相邻O 原子之间争夺电子,因而过渡态较为不稳定,相对自由能高达50.95 kcal/mol。而第三次脱水形成的TS3 的碳正离子中C(4)位空轨道在相邻π-π共轭双键作用下存在离域效应,因而过渡态TS3 较TS1 更稳定一些,其相对自由能为44.61 kcal/mol。第二次脱水为双羟基间脱水,C(1)位羟基的H 容易脱去形成C=O,形成的过渡态TS2 的碳负离子较稳定,且溶剂中H3O+的正电荷能与碳负离子及多个溶剂H2O分子形成较多氢键作用而较大程度的分散正电荷,因而本步基元反应所需活化能较低,过渡态TS2 的相对自由能仅为33.82 kcal/mol。显性溶剂模型中果糖脱水制备5-HMF 反应的控速步骤为呋喃果糖的第一次分子内脱水,其过渡态结构的能量由呋喃果糖C(2)位OH 质子化脱水后形成的碳正离子与溶剂环境中OH-的稳定性决定。实验研究表明,Brønsted 酸催化剂可显著地促进该反应的进行,正是由于酸性环境中控速步骤的亲电进攻更容易发生,且过渡态结构中碳正离子与OH-的稳定性在酸性溶液环境中稳定较高。
3 结 论
本研究采用色散修正密度泛函理论(DFT-D)方法比较研究了亚临界水中隐性和显性溶剂模型描述的葡萄糖异构化为果糖及果糖脱水制备5-HMF的反应机理,根据显性溶剂模型的过渡态结构揭示了Brønsted 酸碱催化剂对不同基元反应促进或抑制的根本原因,并与实验结果相一致。得到以下主要结论:
显性溶剂模型中溶剂H2O 分子能够作为质子传递型溶剂参与到反应中并在质子迁移过程起催化作用,因而显性溶剂模型中葡萄糖异构化为果糖及果糖脱水制备5-HMF 两个反应均表现出比隐性溶剂模型中更低的活化能,更接近真实反应的情况。
显性溶剂模型中葡萄糖异构化为果糖反应放热5.26 kcal/mol,控速步骤为开环葡萄糖的醛-烯醇异构化,所需活化能为33.89 kcal/mol,由于Brønsted碱催化剂很容易提取C(2)位的α-H,且形成的碳负离子在碱性溶液中较为稳定,因而具有较好的催化效果。
显性溶剂模型中果糖脱水制备5-HMF 反应放热12.93 kcal/mol,控速步骤为呋喃果糖的第一次分子内脱水,所需活化能为50.95 kcal/mol,由于碳正离子与溶剂环境中的OH-在酸性溶液环境中稳定较高,因此,Brønsted 酸能够高效催化果糖脱水制备5-HMF 反应。
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
石油石化绿色低碳(2020年2期)2020-12-31
临床肝胆病杂志(2020年1期)2020-12-20
石油石化绿色低碳(2019年6期)2019-01-14
电脑知识与技术(2018年3期)2018-03-21
消费导刊(2017年24期)2018-01-31
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
应用化工(2015年3期)2015-04-01
中学语文(2015年27期)2015-03-01
