
硅铝比对CaO-SiO2-Al2O3熔体微观结构的影响分析
2021-09-03 08:55:56马芳茹买尔哈巴阿不都热合曼王建江王学斌
洁净煤技术 2021年4期
马芳茹,魏 博,买尔哈巴·阿不都热合曼,王建江,李 显,王学斌
(1.新疆大学 煤炭清洁转化与化工过程重点实验室,新疆 乌鲁木齐 830000;2.华中科技大学 煤燃烧国家重点实验室,湖北 武汉 430074;3.西安交通大学 能源与动力工程学院,陕西 西安 710049)
0 引 言
煤炭作为我国不可替代的基础能源,发展和改善洁净煤技术已经成为我国研究学者的重要课题[1]。煤气化技术是现代煤化工发展的龙头技术之一,为降低污染、实现能源可持续发展提供了重要支持[2-3]。煤灰成分复杂、元素多变,一般分为SiO2、Al2O3、TiO2等酸性成分和CaO、MgO、Na2O、K2O等碱性成分,各成分含量直接决定了煤灰的熔融特性和黏温特性[4]。煤灰的酸碱比和硅铝比是影响煤灰特性的关键因素[5-6]。我国典型煤种的硅铝比在0.63~3.50[7],通过在煤中加入添加剂或采用混煤等方法,改变煤灰硅铝比来调节煤灰的熔融特性,是改善煤高温转化过程中带来灰渣问题的重要手段[8]。刘峰等[9]对比分析了不同硅铝比下的新疆气化煤、山东枣庄煤、陕蒙气化煤3种典型煤种,认为硅铝比在2.5左右的煤渣流动特性和黏温特性较好,在一定程度上保护气化炉耐火砖。孟莹等[10]研究了硅铝比对煤灰热膨胀性的影响,研究表明硅铝比高的煤灰有较明显的膨胀性,灰渣表面气孔大。臧卓异[7]从熔融矿物质角度研究不同硅铝比下的碱性氧化物对煤灰熔融特的影响,发现随着硅铝比增大,碱性氧化物对煤灰熔融特征温度的改善效果更加明显。Liu等[11]采用X射线衍射(XRD)和扫描电子显微镜(SEM)研究了合成的煤灰体系并分析煤灰中熔融颗粒微观结构、矿物组成、形貌等变化,证明煤灰的灰熔融温度随硅铝比的增加而降低。但上述研究多从宏观角度分析硅铝比的影响,从体系聚合度、氧的分布等方向的理论研究报道较少,缺乏微观结构对宏观性质改变的理论机理。
第一性原理(First Principle)是从量子力学理论出发的计算方法,根据原子核和电子相互作用的原理及其基本运动规律求解薛定谔方程,进而得到计算物质的基态性质[12]。分子动力学(Molecular dynamics)是模拟体系的动力学演化过程,给出微观量与宏观可观测量之间的关系[13]。模拟成功的关键是体系势函数,可通过第一性原理计算[14]。“第一性原理分子动力学”在煤灰等硅酸盐体系已有应用,得出体系微观结构和宏观量之间关系的准确性已被验证[15-16]。
为了能够具体分析硅铝比的变化对煤灰体系微观结构的影响,得出宏观性质改变的理论机理,本文基于第一性原理,采用分子动力学模拟方法,在1 600 ℃ 下研究由Si、Al、Ca所组成的CaO-SiO2-Al2O3三元体系[17],详细分析了硅铝比从0.5升至3.1 时体系径向分布函数(RDF)、平均配位数(CN)、平均键长、聚合度、氧的分布等微观结构的变化规律,从微观结构解释宏观性质的变化,以期为通过改变煤灰硅铝比调节煤灰熔融特性提供理论参考。
1 计算方法
1.1 势函数确定
模拟体系为CaO、SiO2、Al2O3,在1 600 ℃、0.1 MPa 下研究3种氧化物制备的合成煤灰在熔融状态下的微观结构。保证体系CaO、SiO2与Al2O3总和不变,将硅铝比(SiO2与Al2O3的摩尔比)由0.5升至3.1,模拟体系离子数保持在4 000左右,初始构象为随机构象,体系密度采用经验公式计算[18],具体体系粒子数及密度见表1。

表1 体系粒子数分布
势函数的选取直接影响体系模拟结果的正确性,对于硅酸盐体系,较常见的是Born-Mayer-Huggins(BMH)双体势函数,其正确性已被验证[19-20]。由于 BMH 势参数是离子型两体间势,模拟过程中体系应为熔融态,使体系被视为完全离子溶液,因此模拟体系温度和组分通过Factsage相图选取。BMH势函数的形式[20]为
(1)
其中,U(rij)为粒子i、j的原子间势;rij为粒子i、j之间的距离,m;qi和qj为粒子i、j的有效电荷,C;Aij和Bij为粒子i、j之间的排斥常量;Cij为范德华力常量;r为模拟盒子边长,nm。不同粒子与氧离子间势能参数见表2[21]。

表2 BMH势函数离子间势能
模拟过程中采用NVT系综,使用Parrincllo-Rahman方法[22]进行温度控制、Nosé方法[23]进行压力控制,对于长程库仑力的计算采用Ewald求和方法。在 4 000 K下运行40 000步,使体系中原子充分混合以消除初始分布状态所带来的应力,随后运行64 000步使温度降至1 873 K,最后在1 873 K下驰豫52 000步使体系达到平衡状态。在平衡状态下得到体系各粒子空间坐标,通过对坐标的分析得出体系的微观结构特征。
1.2 径向分布函数和平均配位数
径向分布函数(Radical distribution function,RDF)的物理意义为在某一个粒子距离为r~Δr的另一种粒子的平均数量,第1峰位横坐标反映2个粒子之间的平均键长,从而得出粒子间成键的变化趋势,计算公式[24]如下:
(2)
其中,gij(r)为粒子i、j之间的RDF;nij(r)为以任意一个i粒子为中心,在与其距离为r~Δr的j粒子数量;Ni、Nj为粒子i、j的个数;V为体系总体积,m3。平均配位数(Coordination number function,CN)直接反映体系的结构变化,是衡量熔体结构的重要指标。CN通过对RDF的第1峰谷位进行积分计算得出,公式[25]如下:
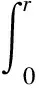
(3)
式中,Nij(r)为粒子i、j间的平均配位数;xj为粒子j的平均数密度。
2 结果与分析
2.1 径向分布函数和平均配位数
为了验证模拟结果的准确性,以文献中常见SiO2/Al2O3=1.5时的计算结果为例,在1 600 ℃下得出各粒子之间的RDF如图1(a)所示,第1峰位对应的横坐标即为各粒子间的平均键长,Si—O、Al—O、Ca—O和O—O的平均键长分别为1.61×10-10、1.75×10-10、2.32×10-10、2.62×10-10m,与文献[26-27]基本一致。图1(b)为各粒子间的CN,其中曲线斜率趋于0时的平滑状态,表示该粒子间存在较稳定的配位结构,对应的纵坐标即为粒子间的平均配位数[17],可以看出,Si—O、Al—O存在稳定的配位结构,其他各粒子间配位数不稳定,说明在煤灰体系中Si、Al为网络形成体,Si—O、Al—O构成了煤灰的基本网络结构[18]。 Si—O、Al—O配位数分别为4.08和4.32,与文献[28]基本一致,验证了模拟结果的准确性。

图1 SiO2/Al2O3=1.5时各粒子间RDF和CNFig.1 RDF and CN between particles when SiO2/Al2O3 is 1.5
2.1.1径向分布函数
1 600 ℃下SiO2/Al2O3由0.5升至3.1时,Si—O和Al—O的RDF如图2所示。由图2(a)可以看出,Si—O间RDF第1峰位不随SiO2/Al2O3的增大而变化,均保持在1.61×10-10m,证明体系中Si—O较稳定,且平均键长为1.61×10-10m。但峰尖逐渐降低,第1峰位越尖锐,表明Si和O的相对距离在该位置存在数量多且较为集中,随着SiO2/Al2O3增大,更多Si—O受到Al的影响,增加了体系的无序性。由图2(b)可知,Al—O平均键长为1.75×10-10m,随着SiO2/Al2O3增大,Al—O存在从1.75×10-10m到1.76×10-10m的迹象,分布并不像Si—O规律,表明Al—O没有Si—O稳定,Al—O在煤灰中的结构比Si—O复杂。同时,Al—O第1峰位也随着硅铝比的升高而降低,体系的有序度降低,变化趋势与Si—O一致。

图2 不同SiO2/Al2O3下Si—O、Al—O的RDF Fig.2 RDF between Si—O and Al—O at different SiO2/Al2O3
2.1.2平均配位数
0.1 MPa、1 600 ℃下SiO2/Al2O3由0.5增至3.1时,Si—O和Al—O的平均配位数如图3所示。在合成的煤灰体系中,仅有网络形成体Si、Al出现似平台状的配位情况,故在此仅分析Si—O、Al—O的配位分布。由图3(a)可知,随着SiO2/Al2O3增大,Si—O平均配位数由4.12降至4.03,Si—O键断裂,Si4+对O2-的吸引力降低。同时,图中出现稳定的平台,说明Si—O以[SiO4]4-四配位结构存在且数量较多、较为稳定[18]。由图3(b)可以看出,随着SiO2/Al2O3从0.5增至3.1时,Al—O平均配位数由4.4降至4.32,平均配位数略大于Si—O,且Al—O 配位以[AlO4]5-四配位结构为主,有少量Al—O键断裂,[AlO4]5-网络结构被破坏。Al—O的配位平台倾斜程度高于Si—O,说明[SiO4]4-四配位结构比[AlO4]5-更稳定。
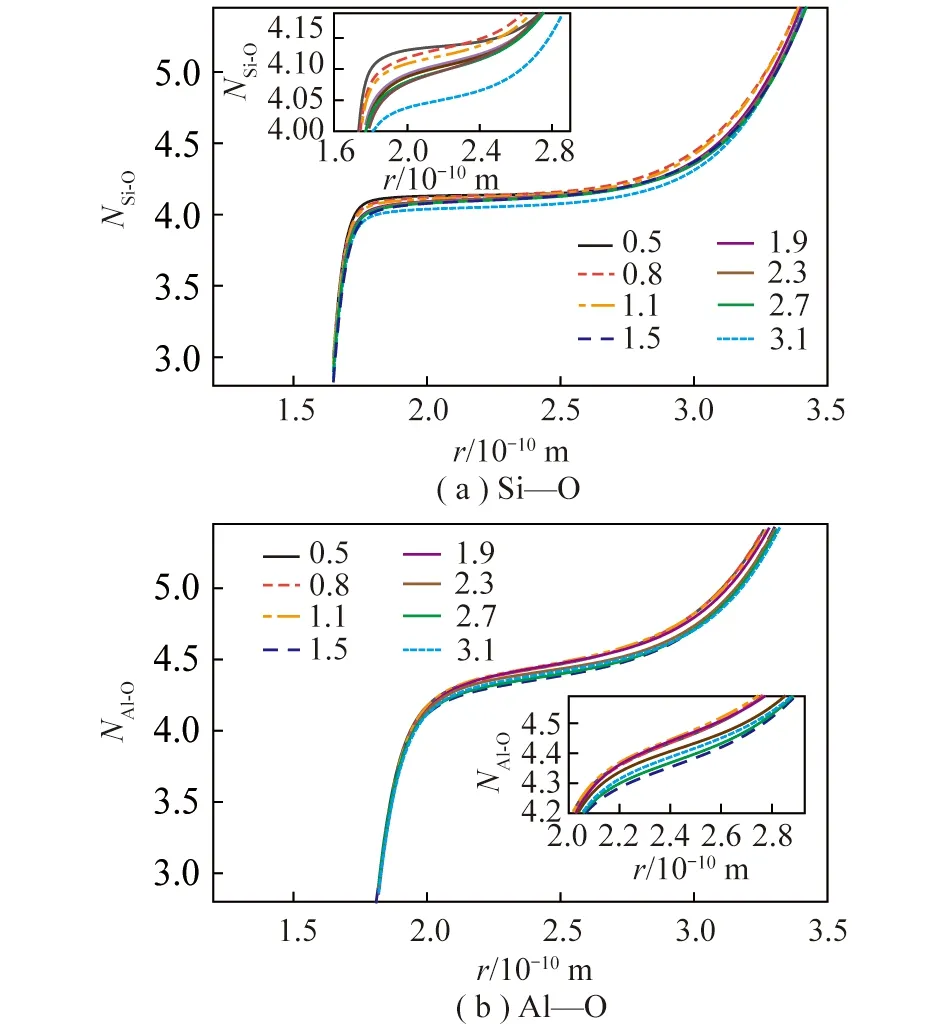
图3 不同SiO2/Al2O3下Si—O和Al—O的CN Fig.3 CN between Si—O and Al—O at different SiO2/Al2O3
2.2 聚合度
硅酸盐体系中常用Qn表示体系的聚合度,其中n为桥氧键的个数[16],Qn可分为Q0、Q1、Q2、Q3、Q4、Q5。图4为1 600 ℃下不同SiO2/Al2O3时Qn的分布情况。随着SiO2/Al2O3增加,聚合度高的Q4、Q5逐渐降低,生成聚合度较低的Q1、Q2、Q3,体系逐渐解聚,由于体系中Q0含量低于0.1%,不予讨论。合成煤灰体系中,Si、Al作为煤灰体系的网络形成体,主要由[SiO4]4-和[AlO4]5-构成煤灰的网络结构。体系聚合度高的Q4从60%降至37%,Q5由17%左右降到7%左右,大量[SiO4]4-和[AlO4]5-被破坏,聚合度低的Q3升高14%左右、Q2升高11%左右。煤灰体系的聚合度降低,无序性升高,网络结构被破坏。随着硅铝比增加,煤的硅酸盐网络结构更易解聚,聚合度的降低使煤渣易产生低温共熔体,表现为煤灰熔融性降低。臧卓异[7]研究表明,硅铝比的增加导致煤灰聚合度降低,高聚集的复杂网络结构变为简单结构,使加入的其他碱性金属氧化物更易代替煤灰硅酸盐中的网络形成体,因此硅铝比的增加更易改变煤灰熔融温度。

图4 不同SiO2/Al2O3下Qn分布Fig.4 Qn distribution under different SiO2/Al2O3
2.3 氧的分布
硅酸盐体系中,氧离子可分为三配位氧(Ot)、桥氧(Ob)、非桥氧(Onb)、自由氧(Of)等4种情况[29],桥氧以Al—O—Al、Al—O—Si、Si—O—Si三种形式存在,三配位氧以O(Al,Al,Al)、O(Al,Al,Si) 两种形式存在[30]。SiO2/Al2O3从0.5增至3.1 时氧的分布如图5所示。随着SiO2/Al2O3增加,Ot和Ob含量降低,Onb含量增加显著。体系中Al3+的减少导致Ot结构中的O(Al,Al,Al)、O(Al,Al,Si)结构开始分解成结构较为简单的Ob和Onb。随着SiO2/Al2O3增加,Ob含量从72%左右降至65%,Ot含量从15%左右降至2%,体系网络结构被破坏,解聚生成的Onb含量由13%升至31%左右。随着硅铝比增加,合成煤灰体系网络结构变得松散,疏松的网络结构导致体系在高温下更易塌陷和瓦解,宏观表现为煤灰的熔融温度降低。这与Liu等[12]研究得到的煤灰灰熔融温度随硅铝比增加而降低的结论一致。

图5 不同SiO2/Al2O3下氧的分布Fig.5 Oxygen distribution under different SiO2/Al2O3
3 结 论
1)随着SiO2/Al2O3增加,Si—O键较稳定,平均键长为1.61×10-10m且以四配位为主;Al—O的平均键长为1.75×10-10m,随着SiO2/Al2O3增加可变化至1.76 ×10-10m;Si—O、Al—O均有较稳定的配位结构,配位数随着SiO2/Al2O3增加而略降,煤灰体系中Si、Al为网络形成体,构成了基本的网络结构。
2)SiO2/Al2O3从0.5增至3.1时,合成煤灰体系中,聚合度高的Q4、Q5逐渐降低,生成聚合度较低的Q1、Q2、Q3,其中Q4、Q5分别降低23%、10%左右,Q3、Q2分别升高14%、11%左右。大量[SiO4]4-和[AlO4]5-被破坏,体系逐渐解聚,有序性降低,表现为煤灰熔融性降低,更易产生低温共熔体。
3)1 600 ℃下SiO2/Al2O3从0.5增至3.1时,Ot和Ob含量逐渐降低,Onb含量增加显著,其中,Ob含量降低7%左右,Ot含量降低13%左右,Onb含量升高18%左右。体系网络结构遭到破坏,疏松的网络结构导致体系在高温下塌陷和瓦解,使煤灰的熔融温度降低。
猜你喜欢
煤质技术(2024年1期)2024-03-18 09:37:06
食品与发酵工业(2021年14期)2021-08-02 12:47:08
煤化工(2021年3期)2021-07-14 07:25:30
陶瓷(2021年5期)2021-06-29 08:07:16
四川水泥(2020年10期)2020-10-27 06:34:12
山东化工(2018年10期)2018-06-07 04:33:33
安徽化工(2016年5期)2016-02-27 08:25:04
化工进展(2015年6期)2015-11-13 00:31:59
华东理工大学学报(自然科学版)(2015年1期)2015-11-07 09:15:47
华东理工大学学报(自然科学版)(2015年1期)2015-11-07 09:15:47
