
TRPC6介导的钙离子紊乱通过激活STAT3促进肾小管上皮细胞损伤后炎症反应及肾间质纤维化*
2021-09-01 08:30练飞鸿王雯倩
中国病理生理杂志 2021年8期
练飞鸿, 付 荣, 王雯倩, 彭 璇△
(1广州中医药大学第一附属医院,广东广州510405;2广州医科大学附属第二医院,广东广州510260)
慢性肾脏病(chronic kidney disease,CKD)的发病率逐年升高,已成为全世界重要的公共卫生问题,而肾间质纤维化(renal interstitial fibrosis,RIF)是各种肾脏疾病进展至终末期肾衰竭的主要病理基础和最终结局。RIF的机制错综复杂,涉及到多细胞、多因素、多途径的共同作用及交互影响[1-2]。研究表明,肾小管上皮细胞(tubular epithelial cells,TECs)在致病因素的作用下极易受到损伤,发生结构及功能的异常。受损的TECs停滞于细胞周期G2/M期,合成、释放大量的炎症因子及促纤维化因子,加强免疫细胞迁移和浸润,使炎症反应持续放大。释放的细胞因子可正反馈作用于TECs自身,促进肾小管上皮-间充质转化(epithelial-mesenchymal transition,EMT)和TECs的继发性损伤,促进肾脏的炎症反应及肾间质成纤维细胞的活化。此外,受损的TECs存在代谢紊乱、线粒体功能受损,进而导致活性氧类释放增多,促进纤维化的发生发展[3-7]。
钙离子(Ca2+)是细胞内重要的第二信使,参与了细胞收缩舒张、增殖分化、递质释放及死亡等多种生命活动的调节。在真核生物体内,细胞通过一系列复杂而严密的机制对Ca2+浓度进行调控。其中,钙池操控性钙通道(store-operated calcium channel,SOC)是调节非兴奋细胞内Ca2+浓度的主要通道,保证信号的正常传递。瞬时受体电位阳离子通道C亚族(tran⁃sient receptor potential cation channel subfamily C,TRPC)是构成细胞膜上SOC的分子基础,该家族包括了TRPC1~7共7个成员,而越来越多的研究显示TRPC6参与了多种疾病的发生发展,其机制可能由于TRPC6基因启动子区域含有活化T细胞核因子(nuclear factor of activated T cells,NFAT)反应元件,参与细胞内Ca2+浓度的持续增高,导致细胞内Ca2+紊乱,进而引起下游疾病相关靶基因的活化[8]。
近年来,越来越多的研究证实TRPC6与肾小球疾病的发病密切相关,足细胞TRPC6基因突变与家族性局灶节段性肾小球硬化症(focal segmental glor⁃nurular sclerosis,FSGS)相关,同时带有该突变基因的患者较快进展至终末期肾病,尽管其机制尚不明确,但一定程度上提示我们可能与TRPC6介导的细胞内Ca2+紊乱相关[9]。既往对TRPC6介导Ca2+的研究多集中于肾小球病变上,其对小管间质损伤的作用及调控机制还所知甚少。
信号转导及转录激活因子(signal transducer and activator of transcription,STAT)信号通路活化参与了细胞多种病理生理过程。作为STAT家族的重要成员,STAT3的活化在细胞增殖、迁移、分化以及炎症/免疫反应疾病的发病过程发挥重要作用。既往研究证实,STAT3磷酸化与肿瘤以及组织纤维化所致的慢性疾病密切相关,参与了肾间质纤维化的启动和维持[10-12]。
本研究利用药物抑制TRPC6介导的Ca2+内流,建立小鼠单侧输尿管梗阻(unilateral ureteric obstruc⁃tion,UUO)[13]模型,观察原代TECs内Ca2+浓度变化及肾小管损伤程度、肾组织炎症细胞浸润、炎症因子水平及肾脏纤维化情况。体外实验通过上调/沉默HK-2细胞TRPC6,研究TGF-β刺激下TECs炎症因子的表达和细胞外基质的合成,探讨其在TECs损伤后炎症反应中的作用及机制。
材料和方法
1 实验动物及实验细胞
雄性SPF级C57BL/6小鼠,8周龄,体重22~25 g,购自广州中医药大学实验动物中心,许可证号为SCXK(粤)2013-0034。所有实验动物均于广州医科大学附属第二医院SPF级实验动物房饲养和繁殖,所有研究工作均遵循广州医科大学《实验动物管理及使用指南》,并由广州医科大学实验动物管理委员会批准(审查编号B2019-050)。HK-2细胞(人肾脏近曲小管上皮细胞)购于ATCC。
2 主要试剂
BTP2和BATPA(Sigma);抗TRPC6、STAT3和p-STAT3Ⅰ抗(Abcam);抗COL1Ⅰ抗(Cell Signaling Technology);抗β-actinⅠ抗(Invitrogen);辣根过氧化物酶(HRP)标记的山羊抗兔IgG(Invitrogen);封闭牛奶和loading buffer(Bio-Rad);PVDF膜(Millipone);Fluo-4 AM(Thermo Fisher Scientific)。
3 主要方法
3.1 小鼠UUO模型的建立 UUO为经典的肾脏纤维化模型,可以在较短时间内导致动物梗阻测肾脏发生肾间质纤维化。本研究选择体重位于22~25 g的雄性C57BL/6小鼠24只,8周龄,随机分为4组:假手术+溶媒组(sham+vehicle),sham+BTP2组,UUO+vehicle组和UUO+BTP2组,每组6只。以1%戊巴比妥麻醉小鼠,仰卧位固定于小鼠板,对小鼠腹部进行消毒,切开左侧腹部暴露并分离左侧输尿管,以丝线靠近肾门端及靠近膀胱端双结扎左侧输尿管,逐层缝合腹膜及皮肤。Sham组打开腹腔后不结扎输尿管,余操作与手术组相同。术后给予常规饲料及饮用水,于术后14 d以颈椎脱臼法处死小鼠,获取肾脏。一部分用于石蜡包埋及切片,行病理染色观察,另一部分置于液氮冷冻后转移至−80℃冰箱保存,用于组织蛋白及核酸的提取。
3.2 小鼠原代肾上皮细胞分离 将假手术组和UUO组小鼠断颈处死,无菌获取双肾,用0.01 mol/L PBS液反复冲洗双肾;去除肾包膜,用无菌剪刀将肾皮质剪碎,吸入15 mL离心管,加入PBS反复多次以清洗组织碎片;离心,去上清,向组织内加入溶解于钙镁平衡盐溶液中的0.25%胰蛋白酶,37℃振荡消化30 min;轻轻吹打分散组织;将组织碎片转移到100目无菌不锈钢丝网上充分研磨,收集液体至200目钢丝网过滤,离心10 min,弃上清,加入45%Per⁃coll分离液25 mL重悬细胞;4℃、28 347×g离心30 min,取近管底的F2层,即为分离纯化的近端肾小管细胞节段,用生理盐水离心洗涤2次,离心5 min,将细胞接种至培养皿中,置于细胞培养箱中培养,观察细胞形态,通过细胞免疫荧光观察细胞CK-18及E-cadherin表达,对肾小管上皮细胞进行鉴定。
3.3 细胞内Ca2+浓度测定 用钙的荧光指示剂Fluo-4/AM孵育细胞30 min;加入含有1%胎牛血清的HBSS孵育40 min后用HEPES液洗涤、重悬细胞,制成约1×108/L的溶液,37℃下培养10 min;于激光共聚焦显微镜专用皿中观察,按[Ca2+]i=Kd×(F−Fmin)/(Fmax−F)将荧光信号值换算成细胞内钙的浓度(nmol/L),其中37℃条件下的Kd值为450 nmol/L。
3.4 Western blot 取小鼠肾组织或HK-2细胞于RIPA裂解液中裂解,超声粉碎30 s;离心15 min后取上清液,使用Bradford蛋白定量法测定蛋白浓度。各样品配至终浓度为2 g/L,加入4×SDS上样缓冲液后沸水水浴使蛋白变性。总蛋白经SDS-PAGE分离、电转至PVDF膜,5%脱脂奶粉溶液室温封闭1 h,加入Ⅰ抗4℃孵育过夜,次日加入Ⅱ抗孵育,化学发光显影。
3.5 real-time PCR 通过基因库(GenBank;http://www.ncbi.nlm.nkh.gov/pubmed/fulltext.htlm)获得PCR引物的基因序列,通过Primer Premier 5.0软件完成引物设计。反应条件为:95℃30 s;95℃15 s,60℃30 s,扩增40个循环。每个样品测3个复孔,分析样品的扩增曲线及熔解曲线,其中ΔCt=Ct目的基因−Ct内参照,ΔΔCt=ΔCt实验组−ΔCt对照组,计算出样品定量结果的平均值。
3.6 细胞培养及质粒转染 HK-2细胞在37℃无菌环境、5%CO2细胞培养箱中培养。使用含有10%胎牛血清的DMEM/F12培养液培养。使用Lipofecta⁃mineTM2000进行TRPC6质粒及TRPC6siRNA转染,转染效率鉴定通过Western blot检测相应蛋白的表达实现。
4 统计学处理
SPSS 19.0统计软件进行数据分析。计量资料用均数±标准误(mean±SEM)表示。两组间比较采用t检验,多组间比较采用单因素方差分析。以P<0.05为差异有统计学意义。
结 果
1 梗阻型肾病模型中TECs内Ca2+浓度增加且伴随TRPC6表达升高
以C57BL/6小鼠为研究对象,建立UUO模型诱导肾脏纤维化,分离鉴定原代肾小管上皮细胞(图1A),使用激光共聚焦技术检测细胞内Ca2+浓度的变化,如图1B所示,与对照组相比,UUO模型组TECs中Ca2+浓度显著增加。Western blot结果显示,与对照组相比,梗阻侧肾脏小管上皮细胞TRPC6表达水平增高,伴随COL1表达增多(P<0.05),见图1C。
2 TRPC6抑制剂减轻UUO模型小鼠肾脏炎症反应及纤维化水平
通过向小鼠腹腔注射BTP2抑制TRPC6通道的活性后观察其对肾脏炎症反应及肾间质纤维化的影响。HE染色结果显示,注射溶媒及BTP2的假手术组小鼠肾组织均未见显著异常;UUO模型小鼠中,注射溶媒后小鼠肾小管显著扩张,TECs细胞扁平,小管间质增宽,伴大量炎症细胞浸润,而注射BTP2则可减轻肾脏上述病理改变,见图2A。进一步检测了小鼠肾组织IL-1β、IL-6及TNF-α3种主要促炎因子的mRNA水平。Real-time PCR结果显示,与sham组相比,UUO模型组小鼠肾组织IL-1β、IL-6和TNF-α mRNA水平显著升高(P<0.05),BTP2可抑制UUO模型小鼠肾组织IL-1β、IL-6和TNF-αmRNA的表达(P<0.05),见图2B。此外,Masson病理染色结果显示,与溶酶模型组比,BTP2干预减少了间质胶原沉积,减轻了肾间质纤维化程度,见图3A。Western blot结果亦显示,BTP2干预可降低肾组织COL1的蛋白水平,结果与染色结果类似,见图3B。
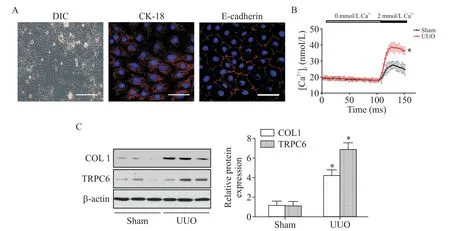
Figure 1.Increasing TECs Ca2+concentration in obstructive nephropathy model was accompanied by increased expression of TRPC6.A:mouse TECs were isolated and cultured and stained with CK-18 and E-cadherin(scale bar=20µm);B:Ca2+concentra⁃tion was detected in mice primary TECs subjected to either sham operation or UUOoperation;C:kidney tissue lysates were subjected to immunoblot analysis for TRPC6 and COL1.Mean±SEM.n=6.*P<0.05 vs shamgroup.图1 梗阻性肾病模型中TECs中Ca2+浓度增加伴随TRPC6表达增多

Figure 2.Pharmalogical inhibition of TRPC6 reduced inflammation in UUOmodel mice.A:representative HEstaining images of kid⁃ney histology showed the morphologic injury and inflammatory cells infiltration and BTP2 injection reduced inflammation caused by obstraction(scale bar=20µm);B:the mRNA expression of IL-1β,IL-6 and TNF-αwas determined by realtime PCR.Mean±SEM.n=6.*P<0.05 vs sham+vehicle group;#P<0.05 vs UUO+vehicle group.图2 BTP2抑制TRPC6减轻UUO模型小鼠肾脏组织炎症反应
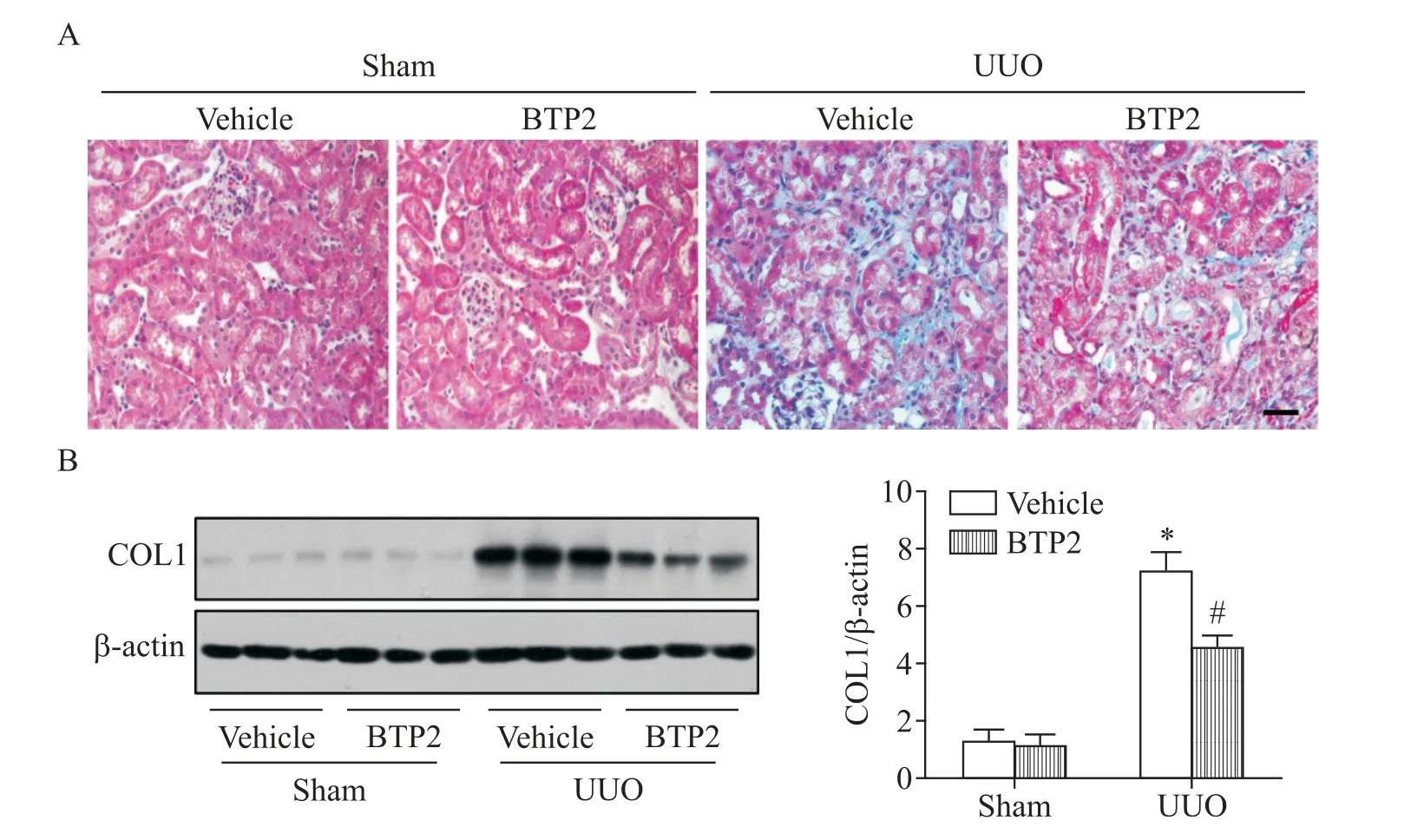
Figure 3.Drug inhibition of TRPC6 reduced renal interstitial fibrosis in UUO model mice.A:representative Masson staining images of kidney histology(scale bar=20µm);B:Western blot analysis of proteins in different groups as indicated.Mean±SEM.n=6.*P<0.05 vs sham+vehicle group;#P<0.05 vs UUO+vehicle group.图3 BTP2抑制TRPC6减轻UUO模型小鼠肾间质纤维化
3 上调TRPC6加重TGF-β作用下HK-2细胞炎症反应及细胞外基质的合成
由于BTP2除了抑制TRPC6通道外,对其他SOC通道也存在一定作用,为了明确TRPC6对肾小管上皮细胞损伤后炎症反应及细胞外基质合成的影响,我们向HK-2细胞转染TRPC6质粒诱导其表达上调,如图4A,与空载体(vector)组相比,转染TRPC6质粒进一步加重了TGF-β刺激下小管上皮细胞炎症因子IL-1β、IL-6及TNF-α 的mRNA表 达(P<0.05);Western blot结果显示,与空载体组相比,细胞转染TRPC6质粒后,TRPC6蛋白的表达增加,同时伴随COL1的表达水平升高,见图4B。

Figure 4.Overexpression of TRPC6 aggravated TGF-β-induced inflammatory response and extracellular matrix synthesis.A:HK-2 cells were transiently transfected with pcDNA3.1-TRPC6 or pcDNA3.1-HA(empty vector)and challenged with TGF-β,and the mRNA expression of IL-1β,IL-6 and TNF-αwas determined by real-time PCR;B:cell lysates were analyzed by immunoblotting with the indicated antibodies.Mean±SEM.n=3.*P<0.05 vs vector group;#P<0.05 vs TGF-β+vector group.图4 上调TRPC6加重TGF-β诱导的HK-2细胞炎症反应及细胞外基质的合成
4 沉默TRPC6抑制TGF-β诱导的HK-2细胞炎症反应及细胞外基质的合成
通过向HK-2细胞转染TRPC6siRNA抑制TRPC6表达,结果显示,在TGF-β刺激下,HK-2细胞炎症因子IL-1β、IL-6及TNF-αmRNA水平较转染scramble siRNA组下调(P<0.05),见图5A。Western blot结果显示,与scramble siRNA组相比,细胞转染TRPC6siRNA后,TRPC6蛋白的表达受到抑制,同时在TGF-β刺激下,HK-2细胞COL1的表达水平下降,见图5B。
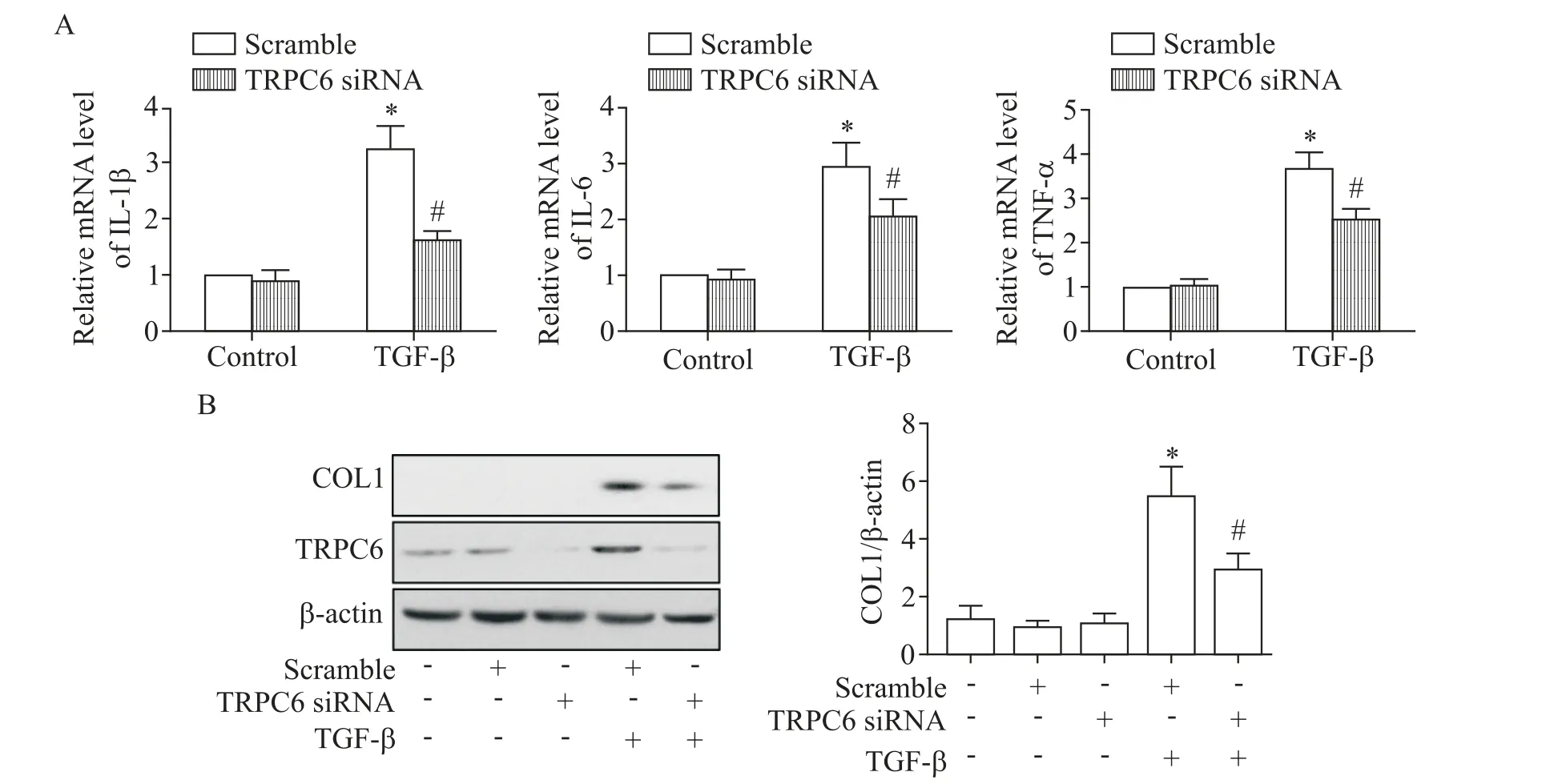
Figure 5.Silencing of TRPC6 alleviated TGF-β-induced inflammation and extracellular matrix synthesis.A:the mRNA expression of IL-1β,IL-6 and TNF-αwas determined by real-time PCR;B:the protein expression of TRPC6 and COL1 was detected by Western blot.Mean±SEM.n=3.*P<0.05 vs scramble group;#P<0.05 vs TGF-β+scramble group.图5 沉默TRPC6减轻TGF-β诱导的HK-2细胞炎症反应及细胞外基质合成
5 沉默TRPC6减弱了TGF-β作用下HK-2细胞STAT3的活化
在UUO模型中检测到STAT信号通路的活化,表现为肾组织p-STAT3表达较sham组增多,而注射BTP2后,p-STAT3水平较溶媒组下降(P<0.05),见图6A。体外实验中,Western blot结果显示,TGF-β刺激下,肾小管上皮细胞STAT3磷酸化水平增加,细胞转染TRPC6siRNA后,STAT3磷酸化水平显著下降(P<0.05),见图6B。
6 高表达TRPC6进一步促进TGF-β作用下HK-2细胞STAT3活化
向HK-2细胞转染TRPC6质粒抑制后检测STAT信号通路活化的情况,结果显示,转染TRPC6质粒进一步促进HK-2细胞中TGF-β诱导的p-STAT3表达,而使用钙离子螯合剂BATPA后,p-STAT3表达被明显抑制,蛋白水平显著降低,见图7。
讨 论
肾间质纤维化的发病机制错综复杂,涉及到多因素、多细胞、多途径的作用,其特征表现为大量细胞外基质的异常沉积,造成肾实质的纤维化,最终导致肾功能的丧失[14]。在致病因素的作用下,肾脏发生炎症反应以清除病原体、修复组织损伤,但级联的炎症反应也是肾间质纤维化的始动因素。TECs是肾实质主要的固有细胞之一,正常情况下,TECs代谢旺盛,担负着重吸收与分泌的功能,具有潜在的增殖及再生能力。越来越多的研究表明,TECs不仅是肾脏病变过程中的“受害者”,更主动参与了肾脏纤维化的形成。受损的TECs停滞于细胞周期的G2/M期,合成、释放大量的炎症因子及促纤维化因子,这些细胞因子加强免疫细胞迁移和浸润,使炎症反应持续和放大,而释放的细胞因子可正反馈作用于TECs自身,促进EMT和TECs的继发性损伤,促进肾脏的炎症反应及肾间质成纤维细胞的活化[15]。我们的研究也显示,在UUO模型中,肾组织发生炎症反应,表现为大量炎症细胞的浸润以及炎症因子合成增多,伴随肾间质纤维化程度较假手术组显著加重。但病理状态下,TECs损伤后生物学行为改变和炎症反应发生发展的机制尚不清楚。
Ca2+是细胞内重要的第二信使,参与了细胞多种生命活动的调节。在真核生物体内,细胞内Ca2+浓度升高主要来源于细胞外钙内流和细胞内钙库的释放,而外钙内流途径主要通过受体门控性钙通道(re⁃ceptor-operated calcium channel,ROC)、电压门控钙通道(voltage-operated calcium channel,VOC)和SOC的调控。在受到相应刺激时,ROC和VOC通道开放,使大量Ca2+短时间内流入细胞内,而SOC则持续产生小量的钙内流,以保证信号的正常传递。TRPC是构成细胞膜上ROC和SOC的分子基础。而越来越多的研究显示TRPC6参与了心血管疾病、肿瘤、呼吸系统等多种疾病的发生发展[8,16-17]。

Figure 6.Inhibition of TRPC6 reduced renal tissue STAT3 activation.A:representative immunoblot analysis of STAT3 and p-STAT3 in UUOmouse kidney extracts;B:relative protein levels of p-STAT3 in HK-2 cells.Mean±SEM.n=3.*P<0.05 vs sham+vehicle group;#P<0.05 vs UUO+vehicle group;△P<0.05 vs scramble group;▲P<0.05 vs TGF-β+scramble group.图6 抑制TRPC6可抑制肾组织及HK-2细胞中STAT3活化

Figure 7.Overexpression of TRPC6 promoted STAT3 activation in HK-2 cells.Mean±SEM.n=3.*P<0.05 vs TGF-β+vector group;#P<0.05 vs TGF-β+TRPC6 plasmid group.图7高表达TRPC6促进HK-2细胞STAT3活化
近年来,关于TRPC6与肾脏疾病的研究取得了显著进展,主要涉及足细胞。该基因突变可导致大量蛋白尿。人类全基因组关联研究从基因水平证明,足细胞Trpc6基因突变与家族性局灶节段性肾小球硬化症相关,同时带有该突变基因患者较快进展至终末期肾病[9]。血管紧张素Ⅱ诱导的高血压肾病模型中,TRPC6基因敲除小鼠蛋白尿显著减少,肾功能水平及肾脏病理改变水平显著优于野生型对照组,并且这种保护作用独立于血流动力的改变[18]。既往对TRPC6的研究多集中于肾小球病变上,其对小管间质损伤所致纤维化的作用及调控机制还所知甚少。我们分离UUO模型及假手术组小鼠原代TECs,显示UUO模型组TECs内Ca2+浓度较对照组显著升高,同时伴随TRPC6蛋白表达水平增高,而药物抑制TRPC6后肾间质炎症反应及纤维化程度得到改善。我们在体外实验中也得到相似的结果,表现为沉默TRPC6表达减轻TGF-β诱导的HK-2细胞的炎症反应及细胞外基质产生,而高表达TRPC6则加重上述反应,提示在损伤因素的作用下,TECs发生Ca2+紊乱,引起肾间质炎症反应及细胞外基质沉积,而Ca2+紊乱可能由TRPC6通道介导。
STAT蛋白家族与机体的炎症及免疫反应密切相关,其中STAT3作为一种转录因子参与调控多种炎症相关基因的表达,在细胞因子合成与释放过程中起着重要作用,参与多种免疫炎症性疾病的发生发展[19-21]。我们的研究观察到,在UUO模型小鼠肾组织中STAT3活化,表现为p-STAT3表达水平增高,同样,在细胞实验中也观察到类似的现象,而通过药物抑制或基因沉默TRPC6表达时,p-STAT3表达受到抑制,高表达TRPC6则促进p-STAT3的表达,而使用钙离子螯合剂处理HK-2细胞后,p-STAT3的表达受到抑制,提示TRPC6可能通过影响Ca2+水平调控TECs STAT3通路,从而促进肾脏炎症反应及肾间质纤维化的发生发展,
综上所述,在梗阻性肾病模型及体外应用TGF-β刺激下,TECs的TRPC6通道活化可引起细胞内Ca2+浓度升高,进一步激活STAT3通路,引起肾脏的炎症反应及肾间质纤维化。
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10
传染病信息(2022年3期)2022-07-15
感染、炎症、修复(2021年1期)2021-07-28
肝博士(2021年1期)2021-03-29
云南化工(2020年11期)2021-01-14
作文成功之路·小学版(2020年6期)2020-07-27
世界科学技术-中医药现代化(2020年2期)2020-07-25
医学研究杂志(2015年6期)2015-07-01
遵义医科大学学报(2014年1期)2014-08-09
中国中医药现代远程教育(2014年13期)2014-03-01
